5 ВЕЛИКИЙ ПРАКТИКУМ. СИНТЕЗ ОРГАНІЧНИХ СПОЛУК
Синтез органічних сполук передбачає більш глибоке вивчення курсу органічної хімії, що базується не лише на вивченні головних теоретичних положень, а і на знаннях та навичках практичної роботи в хімічній лабораторії стосовно техніки безпеки, техніки лабораторних робіт, методів вилучення та очищення органічних речовин, а також властивостей органічних речовин і якісних реакцій на їх функціональні групи.
Таким чином, перші чотири розділи даного посібника є основою для синтезу, виділення та ідентифікації органічних сполук згідно з наведеними методиками. Синтези органічних сполук згруповано за типами реакцій, а наведений у відповідних підрозділах теоретичний матеріал сприяє більш глибокому та цілісному їх вивченню. В кінці кожної методики синтезу наведена коротка характеристика отриманої сполуки, галузі її використання та токсичні характеристики при впливі на людей, тварин та довкілля.
5.1 Алкілування
Алкілування – це реакція введення алкільної групи (Alk –) в органічну сполуку за найбільш поширеною схемою:
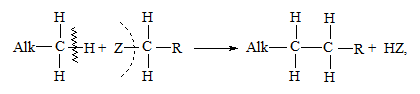
де R = Alk; Z = Н, Hal (Cl, Br, I), OH, NR2 та ін.
Згідно з наведеною схемою, реакцію алкілування можна розглядати як реакцію заміщення атома Гідрогена або атомної групи (Z) на вільний радикал.
В залежності від атома, за яким іде реакція, розрізняють С- , N- , O- та S-алкілування.
Найбільш поширеними алкілуючими агентами є:
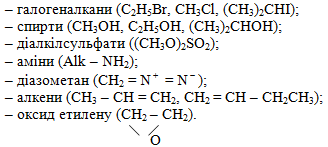
Найбільш поширеним каталізатором при алкілуванні ароматичних сполук є безводний алюміній хлорид (AlCl3).
Наведемо деякі приклади алкілування.
Реакція О-алкілування (реакція А. Вільямсона, 1852 р.) – отримання етерів алкілуванням алкоголятів – (AlkOM) або фенолятів (ArOM) алкілгалогенідами (Alk – Hal) за загальною схемою [41 – 43]:
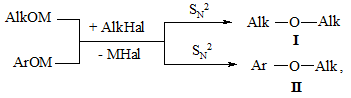
де M = Na, K, Mg, Ag; Hal = Cl, Br, I.
Реакційна здатність Alk – Hal зменшується в ряду: I > Br > Cl, стосовно галогенів і в ряду Alkтрет. > Alkвтор. > Alkперв. стосовно алкільних радикалів.
Реакція проходить за SN2 механізмом з утворенням діалкілових (І) або алкіларилових (ІІ) етерів.
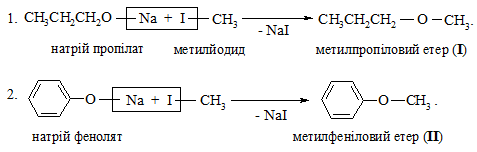
Приклади отримання сполук І та ІІ:
Реакція N-алкілування. В реакцію вступають амоніак, одно- або двозаміщені аміни. При цьому атом Гідрогену, що сполучений із Нітрогеном, заміщується на алкільний (Alk) радикал за схемою:
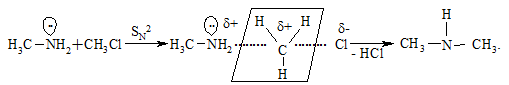
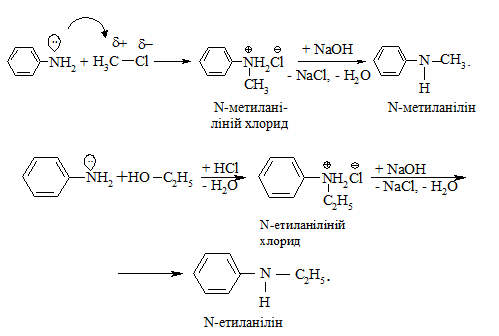
Ароматичні аміни алкілують галогеналканами або аліфатичними спиртами (промисловий метод):
Реакція С-алкілування. Алкілування може проходити за реакцією приєднання молекули, що алкілується (субстрату), до подвійного Карбон – Карбонового зв’язку етиленової сполуки (реагенту) за загальною схемою [41]:
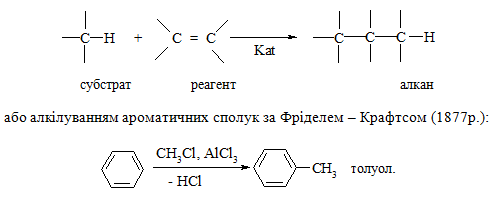
Реакцію, як правило, проводять галогеналканами в присутності алюміній хлориду як каталізатора. Роль каталізатора зводиться до поляризації галогеналкана, тобто до утворення позитивно зарядженого карбонієвого йона:
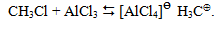
В подальшому реакція заміщення атома Гідрогену ароматичного ядра на алкільний радикал проходить за механізмом електрофільного заміщення (SE) в дві стадії:
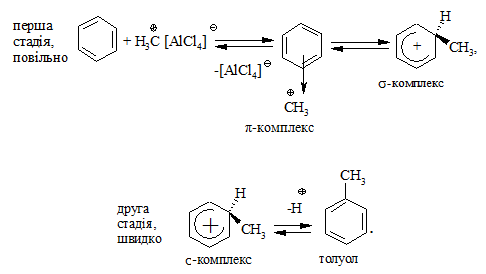
На першій стадії спочатку утворюється нестійкий π-комплекс, який потім переходить у σ-комплекс (карбокатіон). Утворення останнього від-бувається із порушенням ароматичної системи і переходом одного атома Карбону із sp2- в sp3-гібридний стан. Утворення σ-комплексу є найбільш енергетичною стадією реакції, тому позитивний заряд карбокатіона делокалізується за допомогою резонансних структур.
На другій стадії відщеплення протону в присутності сильного нуклеофіла, наприклад AlCl , відбувається швидко з подальшою регенерацією (regeneration) каталізатора:
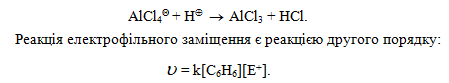
Механізм реакції електрофільного заміщення можна розглянути за допомогою енергетичної діаграми (рис. 5.1). Точка 1 енергетичного профілю відповідає π-комплексу (енергія активації ΔЕ1); точка 2 – σ-комп-лексу (енергія активації ΔЕ2); відщепленню протона і утворенню продукту реакції відповідає енергія активації ΔЕ3. За даною енергетичною діаграмою реакція екзотермічна, що відповідає ΔЕр реакції.
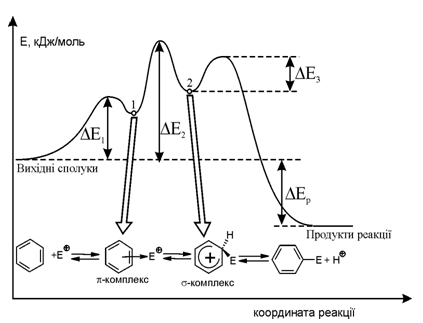
Реакцію S-алкілування можна віднести до реакцій отримання аліфа-тичних та ароматичних тіоетерів (сульфідів) за схемою:
Арени при дії дихлорсульфуру SCl2 в присутності каталізаторів кислот Льюїса вступають в реакцію електрофільного заміщення:
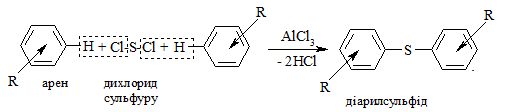
Реакція алкілування дуже широко використовується в промисловості (органічний синтез) для синтезу багатьох органічних сполук (толуол, ксилоли, кумол), синтезу високооктанового палива тощо.
5.1.1 Ізопропілбензол. С-алкілування
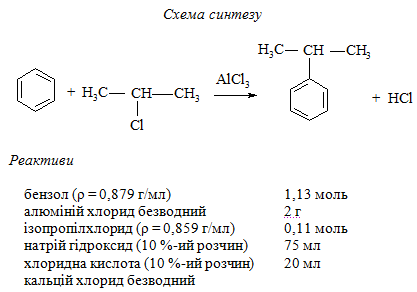
Методика синтезу. Круглодонну колбу об'ємом 300 мл з'єднують за допомогою дворогого форштоса з краплинною лійкою і зворотним холодильником, які закривають хлоркальцієвими трубками. До зовнішнього кінця хлоркальцієвої трубки приєднують скляну трубку, занурену в колбу чи стакан з водою для поглинання хлороводню. Кінець трубки повинен знаходитись на відстані 1 см від поверхні води. В колбу вносять 0,9 моль висушеного бензолу, 2 г безводного подрібненого алюміній хлориду і суміш нагрівають на водяній бані (≈ 80 °С).
В краплинній лійці змішують 0,11 моль ізопропілхлориду і 0,23 моль висушеного бензолу. Отриманий розчин по краплинах вносять в нагріту реакційну масу, після чого колбу витримують на водяній бані (≈ 80 °С) до припинення виділення хлороводню. Хід реакції контролюють за допомогою змоченого у воді індикаторного паперу, який не повинен набувати рожевого забарвлення. Потім реакційну суміш виливають в стакан з льодом і додають 20 мл 10 %-го розчину хлоридної кислоти, переносять в ділильну лійку, відділяють верхню фазу, яка являє собою розчин ізо-пропілбензолу в бензолі, в конічну колбу, промивають його спочатку 10 %-ним розчином натрій гідроксиду (трьома порціями по 25 мл), потім водою до нейтральної реакції і висушують безводним кальцій хлоридом.
Виділення продукту. Розчин фільтрують через складчастий фільтр, переносять в колбу Вюрца, відганяють спочатку бензол з водяним холодильником, а потім ізопропілбензол з повітряним холодильником, відбира-ють ізопропілбензольну фракцію з Ткип = 151 – 153 ºС.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.1.

Характеристика кінцевого продукту. Ізопропілбензол (кумол) – безбарвна рідина, легко загоряється, утворює вибухонебезпечні суміші. Нерозчинна у воді, змішується з діетиловим етером, етиловим спиртом, ацетоном, хлороформом, бензолом, Тпл = –96,3 °С, Ткип = 52 °С, d420 = 0,8618, nd20 = 1,4913.
Токсичність. Отруйний, при інгаляції викликає гострі і хронічні ураження кровотворних органів. ГДК р.з. = 50 мг/м3, ЛД50 = 15,3 мг/кг.
Використання. Як розчинник лаків і фарб, як високооктанова добавка до авіаційних бензинів і вихідна речовина при виробництві фенолу і ацетону.
5.1.2 Діоксан. О-алкілування
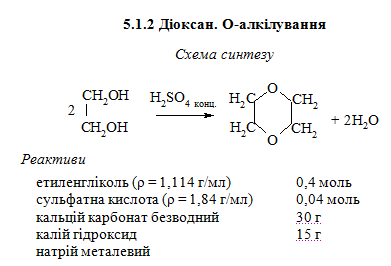
Методика синтезу. В круглодонну колбу, що оснащена дефлегматором з термометром та похилим холодильником, поміщають 0,4 моль етиленгліколя та 2,4 мл концентрованої сульфатної кислоти. Реакційну суміш обережно (у витяжній шафі!) нагрівають до кипіння на електричній плитці із азбестованою сіткою. Через деякий час в температурному інтервалі 84 – 102 °С отримують продукт реакції. Відганяти діоксан потрібно повільно, а нагрівання потрібно закінчити, як тільки реакційна суміш починає темніти та спінюватись (Т ≈ 102 °С). Відігнаний діоксан переносять у ділильну лійку і добавляють 15 г кристалічного калій карбонату (до утворення двох шарів).
Виділення продукту. Верхній шар, що являє собою діоксан, відділяють в ділильній лійці та висушують в плоскодонній колбі ємністю 100 мл спочатку прожареним калій карбонатом, а потім обробляють калій гідроксидом для видалення ангідриду оцтової кислоти, що утворюється як побічний продукт реакції. Висушений продукт переносять в колбу Вюрца та переганяють над невеликими шматочками металевого натрію, відбирають фракцію в інтервалі Ткип = 100 – 103 °С.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.2.

Характеристика кінцевого продукту. Діоксан – безбарвна горюча рідина з слабким запахом. Змішується з водою, спиртом, етером, Тпл = = 11,8 °С, Ткип = 101,32 °С, d420 = 1,034, nd20 = 1,4224.
Токсичність. За токсичними характеристиками подібний до аліфатичних етерів. ГДКр.з. = 10 мг/м3, ЛД50 = 37 мг/кг.
Використання. Використовується при очищенні нафтових масел та як розчинник для ацетилцелюлози, смол, каучуків, мінеральних та рослинних олій.
5.1.3 N,N-Діетиланілін. N-алкілування
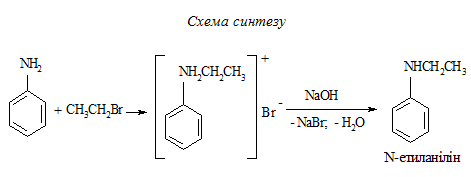
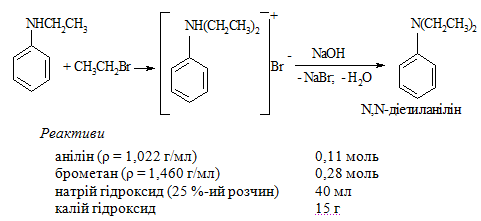
Методика синтезу. В круглодонній колбі об'ємом 100 мл, обладнаній зворотним холодильником, кип'ятять 0,11 моль перегнаного аніліну з 0,14 моль брометану протягом 2 год (до затвердівання реакційної маси). Електричну плитку замінюють на водяну баню для охолодження реакційної маси. До охолодженої реакційної суміші невеликими порціями при постійному перемішуванні додають 20 мл 25 %-го розчину натрій гідроксиду.
Утворений N-етиланілін відділяють за допомогою ділильної лійки (верхній шар) і знову переносять в реакційну колбу. Додають 0,14 моль брометану і кип'ятять на електричній плитці з азбестовою сіткою до за-твердівання реакційної маси. Отриману сіль розчиняють в 35 – 40 мл во-ди, розчин переливають в стакан і кип'ятять впродовж кількох хвилин для видалення брометану, який не вступив в реакцію. Увага! Кип'ятіння проводити у витяжній шафі! Потім розчин охолоджують до кімнатної температури і при перемішуванні додають до нього невеликими порціями 20 мл 25 %-го розчину натрій гідроксиду.
Виділення продукту. Отриманий N,N-діетиланілін відділяють за допомогою ділильної лійки (верхній шар) і переносять в конічну колбу об'ємом 100 мл, висушують за допомогою калій гідроксиду, фільтрують через складчастий фільтр в колбу Вюрца і переганяють, відбирають фракцію з Ткип = 214 – 216 ºС.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.3.
Характеристика кінцевого продукту. N,N-діетиланілін – масляниста рідина світложовтого кольору. Розчиняється в спирті, діетиловому етері, нерозчинна у воді, Тпл = 38,8 °С, Ткип = 215,5 °С, d420 = 0,935, nd20 = 1,5411.
Токсичність. ЛД50 = 290 мг/кг (для білих пацюків), ЛД50 = 500 мг/кг (для білих мишей).
Використання. В виробництві трифенілметанових барвників, для отримання діетиламіну.
Контрольні запитання
1. Яку реакцію називають реакцією алкілування? Наведіть загальну схему реакції.
2. Назвіть найбільш поширені алкілуючі агенти.
3. Якою реакцією (С-, N-, O- чи S-алкілуванням) отримують етери з ал-коголятів та фенолятів? Наведіть рівняння реакції отримання етилбутилового етеру з бутилового спирту.
4. Які речовини використовують як алкілуючі агенти в реакції N-алкі-лування? Які речовини є субстратами в цій реакції?
5. Якою реакцією отримують алкілзаміщені бензолу? Наведіть схему реакції отримання етилбензолу з бензолу.
6. Які алкілуючі агенти та каталізатори застосовують при С-алкілу-ванні ароматичних сполук?
7. За якими механізмами протікають реакції С-, N- , O-алкілування?
8. Які продукти отримують реакцією S-алкілування?
9. Який алкілуючий агент використовують при отриманні ізопропілбензолу з бензолу? Наведіть схему та механізм реакції.
10. Який алкілуючий агент використовують при отриманні N,N-діетил-аніліну з аніліну? Наведіть схему та механізм реакції.
5.2 Ацилювання
Ацилювання – це введення ацильного радикала –C(=O)R в органічну сполуку реакцією заміщення як найбільш вивченою та доступною. В зале-жності від того, біля якого атома заміщується рухомий Гідроген на ацильний радикал розрізняють C-, N-, O- та S-ацилювання. Наприклад, реакція C-ацилювання відбувається за загальною схемою [41, 44, 45]:
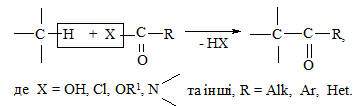
За активністю найбільш поширені ацилюючі засоби можна розмістити в ряд:
Реакція С-ацилювання. Ароматичні вуглеводні ацилюються реакцією Фріделя – Крафтса подібно до реакції алкілування ароматичних сполук:
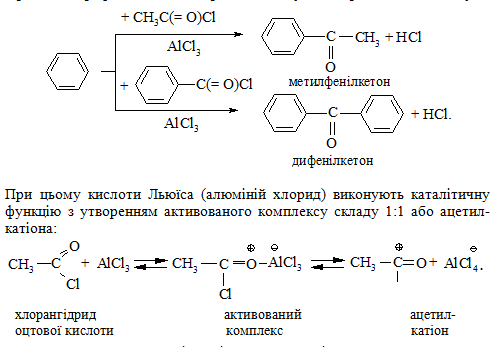
В подальшому реакція заміщення атома Гідрогена в ароматичному ядрі на ацетилкатіон проходить в дві стадії за механізмом електрофільного заміщення SE:
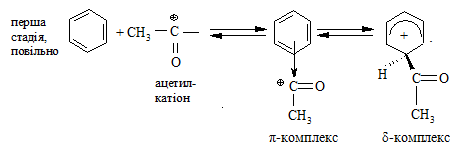
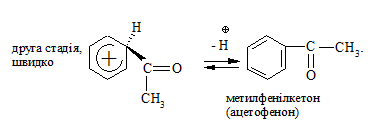
Реакція С-ацилювання ароматичних сполук проходить за SE механізмом подібно до реакції С-алкілування, включаючи повільну, лімітуючу (першу стадію утворення σ-комплексу) та швидку (другу стадію утворення кінцевої сполуки) стадії реакції.
Реакція О-ацилювання. Прикладом О-ацилювання органічних спо-лук є реакція естерифікації спиртів, яка відбувається при їх взаємодії з мінеральними та карбоновими кислотами:
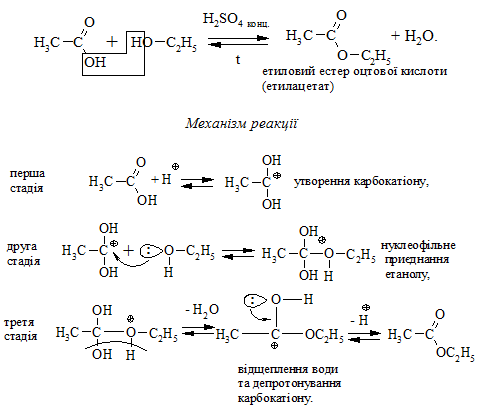
За механізмом – це бімолекулярна реакція приєднання, яка має ряд особливостей:
- реакція рівноважна, тому для одержання кінцевого естеру з макси-мальним виходом необхідно створювати значний надлишок вихідних речовин або виводити із реакційного середовища воду та кінцевий естер;
- швидкість реакції для спиртів різної будови змінюється у ряду: νперв. : νвтор. : νтрет. = 20 : 3 : 1. В першому наближенні це можна пояснити стеричними перешкодами;
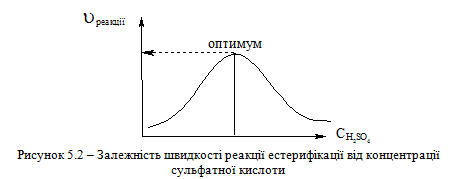
швидкість реакції визначається оптимальною концентрацією сульфатної кислоти (рис. 5.2). Значна концентрація кислоти приводить до про-тонування молекули спирту R – OH + H⊕ RO⊕H2 і відповідно до силь-ного сповільнення реакції. Мала концентрація кислоти значно збільшує кількість води в реакційному середовищі, що зміщує рівновагу у сторону вихідних сполук.
Естери карбонових кислот можна отримати також при взаємодії спиртів з галогеноангідридами або ангідридами кислот:
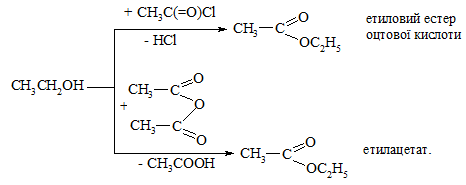
У цьому випадку говорять про ацилювання спирту, оскільки спирт нуклеофільно атакує ангідрид або галогеноангідрид за схемою:
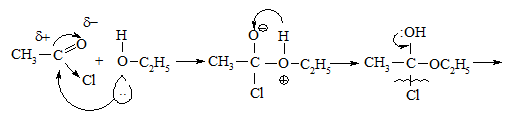
Реакція N-ацилювання. Первинні та вторинні алкіламіни легко аци-люються похідними карбонових кислот (ангідридами, хлороангідридами) з утворенням амідів:
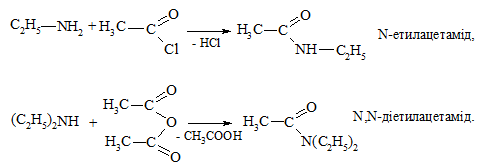
Ацилювання ариламінів, які є менш основними у порівнянні з алкі-ламінами, відбувається при взаємодії ангідридів або хлороангідридів кислот, а у випадку карбонових кислот як ацилюючих агентів потребує жорстких умов перебігу реакції.
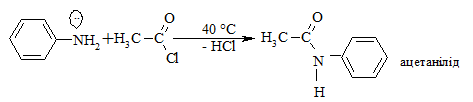
Аміди кислот легко гідролізуються в кислому або лужному середовищі з відновленням (reduction) аміногрупи.
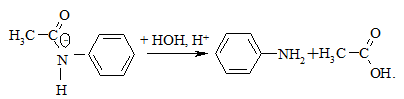
Тому такий підхід використовують в синтетичній практиці для захисту аміногрупи або для зменшення її донорського впливу на ароматичне ядро.
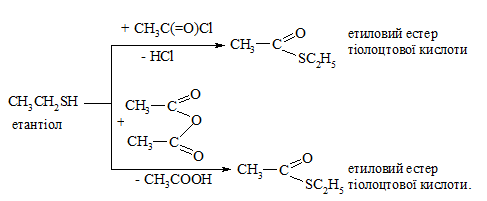
Реакція S-ацилювання. В реакцію вступають етилтіоли (меркаптани) подібно до аліфатичних спиртів за схемою:
5.2.1 Ацетофенон. С-ацилювання
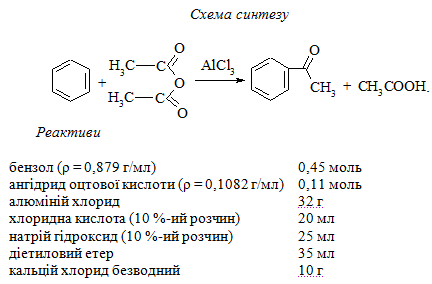
Методика синтезу. Круглодонну тригорлову колбу об'ємом 300 мл, оснащену мішалкою, краплинною лійкою з хлоркальцієвою трубкою і зворотним холодильником, верхній кінець якого закривають хлоркальцієвою трубкою з уловлювачем для хлороводню, поміщають у водяну баню, обладнану термометром.
В колбу вносять 0,45 моль висушеного бензолу і 32 г тонко подрібненого алюміній хлориду. При охолодженні колби в кристалізаторі холодною водою в суміш обережно по краплинах, при інтенсивному перемішуванні, протягом 30 хв за допомогою краплинної лійки додають 0,11 моль оцтового ангідриду. Реакція ацилювання супроводжується виділенням тепла, тому суміш розігрівається і відбувається інтенсивне виділення хлороводню. Після додання оцтового ангідриду для закінчення реакції колбу нагрівають на водяній бані (80 – 85 ºС) протягом 45 хв.
Виділення продукту. Охолоджену до кімнатної температури реакційну суміш виливають в стакан, який містить 80 мл води з льодом. У випадку утворення осаду основної солі алюмінію його розчиняють при доданні 10 – 20 мл 10 %-го розчину хлоридної кислоти. Далі розчин переносять в ділильну лійку, екстрагують 20 мл етеру, відділяють етерно-бензольний шар в конічну колбу ємністю 100 мл, а водний розчин знову обробляють 15 мл етеру. Об'єднані етерні витяжки промивають у ділильній лійці водою, 10 %-им розчином натрій гідроксиду, знову водою, відділяють в конічну колбу об'ємом 100 мл і висушують за допомогою кальцій хлориду.
Розчин фільтрують через складчастий фільтр в колбу Вюрца. Далі відганяють етер і бензол на водяній бані. Потім замінюють водяний холодильник на повітряний, переганяють ацетофенон і відбирають фракцію з Ткип = 199 – 203 ºС.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.4.

Характеристика кінцевого продукту. Ацетофенон (метилфенілке-тон) – безбарвна, іноді з жовтуватим забарвленням масляниста рідина або крупні легкоплавкі кристали. Нерозчинний у воді, добре розчинний в спирті, етері, хлороформі, бензолі, Тпл = 20,5 °С, Ткип = 202,3 °С, d420 = 1,0281, nd20 = 1,5342.
Токсичність. ГДКр.з. = 5 мг/м3, ЛД50 = 1350 мк/кг. Невідкладна терапія та індивідуальний захист такі ж, як і у випадку з ацетоном.
Використання. Використовується в парфумерії, має снодійні властивості.
5.2.2 Ацетилсаліцилова кислота. О-ацилювання
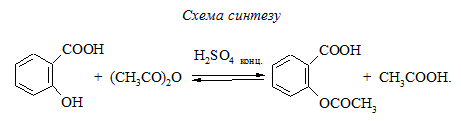
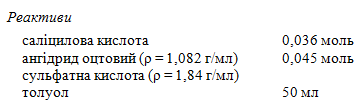
Методика синтезу. Роботу проводять у витяжній шафі! В конічну колбу об'ємом 50 мл, обладнану зворотним холодильником, при слабкому нагріванні вносять 0,036 моль саліцилової кислоти, розчиненої в 0,045 моль оцтового ангідриду і додають три краплини концентрованої сульфатної кислоти. Суміш витримують на водяній бані одну годину при температурі 60 °С і одну годину при температурі 90 °С. Потім реакційну суміш виливають в стакан, періодично перемішуючи скляною паличкою, охолоджують в кристалізаторі холодною водою з льодом.
Виділення продукту. Закристалізований продукт фільтрують під вакуумом на лійці Бюхнера, промивають холодною водою, а потім – невеликою кількістю охолодженого толуолу (≈ 50 мл).
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.5.
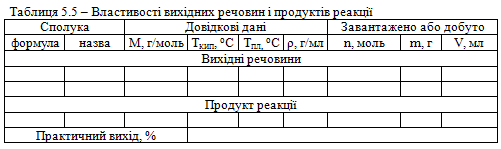
Характристика кінцевого продукту. Ацетилсаліцилова кислота (аспірин) – безбарвна кристалічна речовина. Розчиняється в етиловому спирті, розчинах лугів та карбонатів, погано розчиняється у воді та етері, Тпл = = 136,5 °С, Ткип = 140 °С.
Використання. Як жарознижувальний засіб в медичній практиці.
5.2.3 Бутилацетат
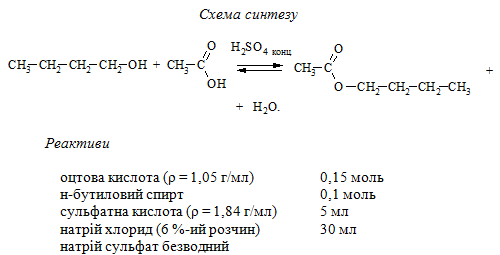
Методика синтезу. В суху плоскодонну колбу на 100 мл вносять 0,15 моль оцтової кислоти при перемішуванні та охолодженні колби під струменем холодної води, а потім невеликими порціями 5 мл концентрованої сульфатної кислоти. За тих же умов повільно (впродовж ≈ 15 хв) до реакційної суміші додають 0,1 моль н-бутилового спирту. Температура реакційної суміші при цьому не повинна перевищувати 40 oС (контролюють за допомогою термометра, зануреного в реакційну суміш).
Плоскодонну колбу з реакційною сумішшю та кип’ятильними камінцями з’єднують із зворотним холодильником та нагрівають на електроплитці протягом 30 хв при слабкому кипінні реакційної суміші.
Виділення продукту. Після завершення реакції суміш охолоджують і додають до неї 30 мл 6 %-го розчину натрій хлориду. Вміст колби переносять до ділильної лійки, струшують і дають відстоятися. Нижній водний шар відокремлюють, а естер, що залишився у лійці, нейтралізують розчином соди (контролюють за допомогою універсального індикаторного паперу), знову відокремлюють і промивають водою.
Естер переносять в суху колбу на 50 мл, висушують реакційну масу безводним натрій сульфатом і переганяють її при атмосферному тиску із колби Вюрца, відбирають при цьому фракцію з Ткип = 124 – 125 °С.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.6.

Характеристика кінцевого продукту. Бутилацетат – безбарвна го-рюча рідина з характерним етерним запахом. Малорозчинний у воді, добре розчиняється в органічних розчинниках, Тпл = 73,5°С, Ткип = 126,1°С, d420 = 0,881, nd20 = 1,3941.
Токсичність. ГДК р.з. = 200 мг/м3. Подразнює слизову оболонку очей та органів дихання.
Використання. Як розчинник для нітроцелюлозних лаків і фарб та екстрагент в парфумерній промисловості.
5.2.4 Ацетанілід. N-ацилювання
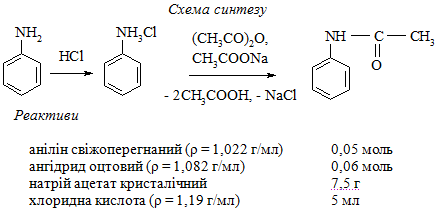
Методика синтезу. В стакан, що містить 250 мл дистильованої води, додають 5 мл концентрованої хлоридної кислоти і при перемішуванні вносять 0,05 моль перегнаного аніліну. Розчин нагрівають на водяній бані до 50 °С і при інтенсивному перемішуванні додають 0,06 моль оцтового ангідриду та одразу ж – розчин, який містить 7,5 г натрій ацетату в 50 мл дистильованої води.
Виділення продукту. Кристалічний осад, який утворився, фільтрують на лійці Бюхнера, промивають невеликою кількістю холодної води (три порції по 50 мл) і висушують.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.7.

Характеристика кінцевого продукту. Ацетанілід – безбарвна кристалічна речовина без запаху. Розчинна в діетиловому етері, хлороформі, етиловому спирті, помірно розчинна у воді, Тпл = 114 °С, Ткип = 303,8 °С, d420 = 1,026, nd20 = 1,3316.
Токсичність. Викликає перетворення гемоглобіну в метгемоглобін і гемоліз еритроцитів.
Використання. Як жарознижувальний засіб в медицині, напівпро-дукт при синтезі барвників і ліків та стабілізатор Н2О2. Біопрепарат у медицині та ветеринарії.
Контрольні запитання
1. Яку реакцію називають реакцією ацилювання? Назвіть найбільш по-ширені ацилюючі агенти.
2. За яким механізмом протікає реакція С-ацилювання ароматичних сполук?
3. Яким чином кислоти Льюїса (AlBr3) активують ацилброміди в реакціях ацилювання ароматичних бензоїдних структур?
4. Наведіть схему та механізм реакції етерифікації пропанової кислоти етанолом.
5. Назвіть умови, за яких проходить реакція етерифікації та її особливості.
6. Які ацилюючі агенти застосовують для отримання естерів реакці-єю О-ацилювання?
7. Наведіть реакцію N-ацилювання. В яких умовах вона протікає і яким чином використовується в органічному синтезі?
8. Як можна захистити аміногрупу в процесі синтезу? Як знімають захист аміногрупи?
9. Які продукти отримують реакцією S-ацилювання? Наведіть приклади.
10. Наведіть схему та механізм реакції синтезу бутилацетату з бутанолу.
5.3 Галогенування
Галогенування – це введення атома галогену в молекулу органічної сполуки за допомогою, як правило, реакцій заміщення або приєднання.
Заміщення атома Гідрогену на галоген проходить за загальною схемою:
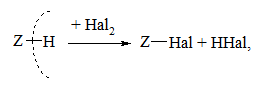
де Z = C, N, S – атоми органічної сполуки.
Багато із реакцій наведеної схеми є іменними. Так, реакція Хелля-Фольхарда-Зелінського – це галогенування карбонових кислот в α-положення при дії галогену та червоного фосфору [46]:
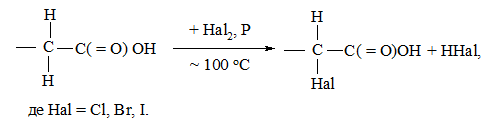
Заміщення групи атомів або атома, відмінного від Гідрогену, на галоген відбувається за схемою:
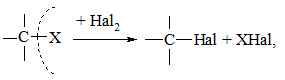
де X = OH, NR2, OSO3R, SO3H та ін.
Приєднання атома галогену до подвійного Карбон-Карбонового зв’язку (С = С-дигалогенування):
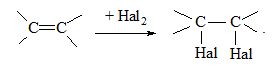
Розглянемо деякі приклади галогенування, яке здійснюється за до-помогою реакцій заміщення та приєднання.
Галогенування алканів під дією галогенів проходить із утворенням моно- та полігалогенопохідних алканів. Загальна схема утворення монопохідних галогеноалканів:
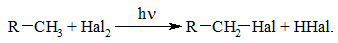
Швидкість реакції в залежності від природи алкану зменшується у ряду: третинний > вторинний > первинний, а в залежності від природи галогену у такій послідовності: F2 (з вибухом) > Cl2, Br2 (опромінення світлом або Т ≥ 300 °C) > I2 (практично не взаємодіє без застосування спеціальних йодуючих агентів, наприклад, (СН3)3С–ОІ).
Хлорування (chlorination) метану відбувається за схемою радикального заміщення (SR):
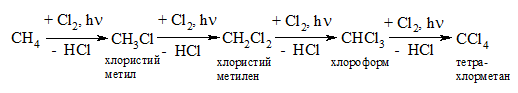
Механізм реакції (SR) ланцюгово-радикальний (М. М. Семенов, Нобелевський лауреат з хімії):

На рисунку 5.3 наведено енергетичну діаграму реакції хлорування метану. Реакція між метаном та хлором проходить при досягненні енергії активації - Е", подальша взаємодія метильного радикала (СН3∙) та молекули хлору відбувається при досягненні енергії активації - Е" з утворенням хлористого метилу та активного радикала хлору.
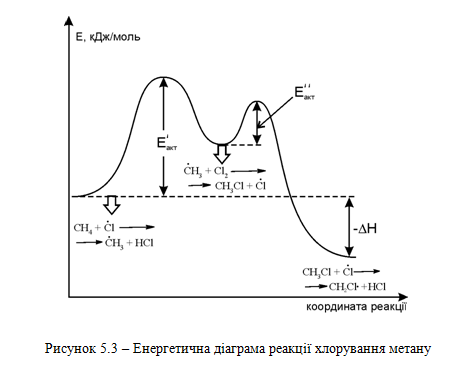
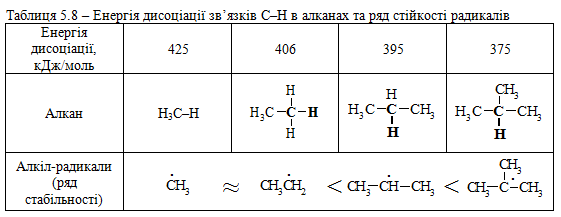
Галогенування різних алканів проходить селективно в залежності від їх будови та стабільності утворених радикалів: третинний > вторинний > первинний. При цьому їх термодинамічна стабільність визначається енергією дисоціації зв’язку С–Н, тобто енергією утворення вільних радикалів та ефективністю делокалізації заряду радикального центра (табл. 5.8).
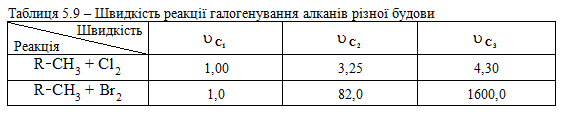
Можливість селективного галогенування закономірно пов’язана із природою галогену (Cl2, Br2) та швидкістю таких реакцій біля первинного (νc1 ), вторинного (νc2 ) та третинного (νc3 ) атома Карбону (табл. 5.9).
У випадку взаємодії (супряження) неспареного електрона із π-електронною системою (алкени, арени) стабільність таких радикалів значно зростає:
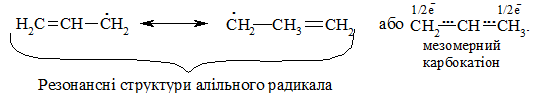
Кінетична стабільність радикалів зумовлена, в першу чергу, просторовими перешкодами, що впливає на їх реакційну здатність. Так, 4-метил-2,6-дитрет-бутилфенол (іонол)
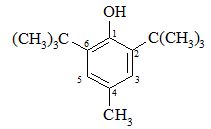
має об’ємні трет-бутильні радикали, що знаходяться у ортоположенні відносно гідроксильної групи. У випадку утворення радикала
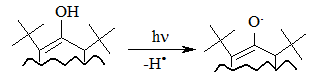
об’ємні трет-бутильні замісники екранують атом Оксигену та заважають перебігу радикальних реакцій по цьому реакційному центру. Виходячи із цього положення, іонол широко використовується у багатьох промислових сумішах (пластмаси, гуми, мастила, масла тощо) як ефективний антиоксидант.
Розглянуті на прикладі реакції галогенування алканів теоретичні положення (природа субстрата та реагента, стабільність радикалів) будуть характерними і для інших реакцій, що відбуваються за радика-льно-ланцюговим механізмом (SR).
Галогенування алкенів проходить з утворенням віцинальних дигалогеналканів:
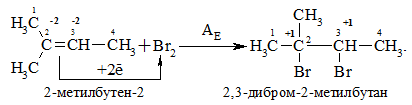
Це – якісна реакція на алкени: бромна вода темно-червоного кольору знебарвлюється у присутності алкену внаслідок утворення дибромгалогеналкану. Реакцію галогенування алкенів можна розглядати як окисно-відновну, в якій алкен окиснюється до дибромгалогеналкану, а молекулярний бром відновлюється до бромід-аніону, що входить до складу 2,2-ди-бром-2-метилбутану.
Швидкість реакції галогенування залежить від природи галогену і зменшується в ряду: F2 (з вибухом) > Cl2, Br2 (кімнатна температура) > I2 (повільно при освітлені, hν), а також від будови алкену:
- чим більше замісників знаходиться біля атомів Карбону подвійного зв’язку, тим активнішим є алкен
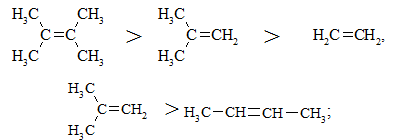
- чим полярнішим є алкен, тим він більш реакційноздатний.
Реакції бромування та хлорування відбуваються, як правило, за АЕ-механізмом:
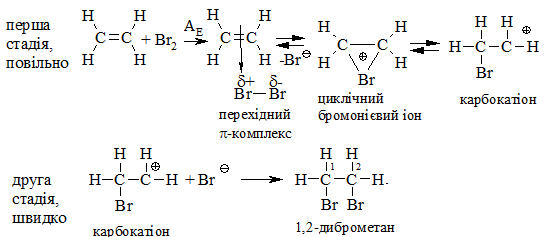
Приєднання галогеноводнів (гідрогалогенування) до алкенів відбувається дуже легко з утворенням галогеноалканів:
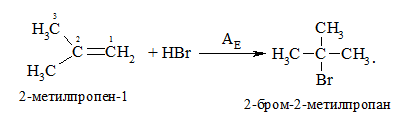
Реакційна здатність галогеноводнів зростає із збільшенням сили кислот у ряду: HF < HCl < HBr < HI.
Галогеноводні у випадку несиметричних алкенів приєднуються за правилом В. В. Марковнікова (1869 р.): атом Гідрогену приєднується до більш гідрогенізованого атома Карбону (до атома Карбону, що має більшу кількість атомів Гідрогену). Така регіоселективність визначається поляризацією подвійного зв’язку в алкенах з утворенням на першій стадії перехідного π-комплексу:
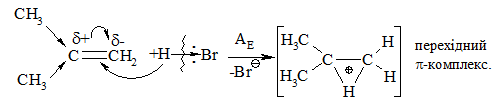
Останній перегруповується до карбокатіонів (І) та (ІІ):
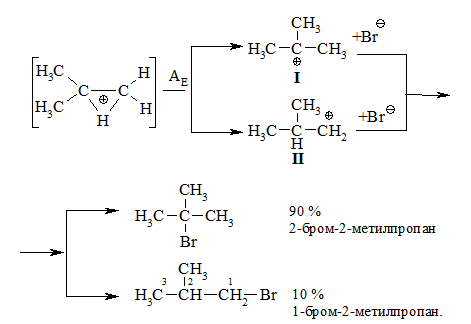
Із двох проміжних карбокатіонів І та ІІ, перший є більш стійким, тому і вихід кінцевого бромалкану у цьому випадку є визначальним (90 %).
При наявності у реакційному середовищі пероксидів реакція проходить не за АЕ-механізмом, а за радикальним механізмом – AR (Хараш, 1933р.):
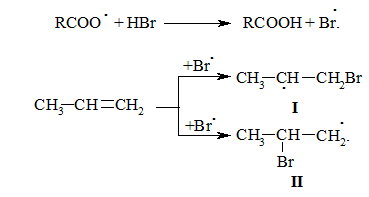
Радикал І є більш стабільним, ніж радикал ІІ, що є визначальним при утворенні кінцевого 1-бромпропану (90 %):
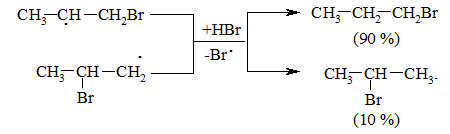
Радикали І та ІІ стабілізуються за рахунок взаємодії із наступною молекулою бромоводню.
Таким чином, наявність в реакційному середовищі пероксидів приводить до зміни механізму реакції та до приєднання галогеноводнів всупереч правилу Марковнікова.
Галогеноарени утворюються при галогенуванні ароматичних сполук хлором або бромом в присутності кислот Льюїса (FeCl3, AlCl3):
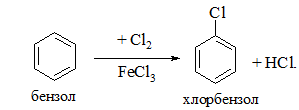
Реакція проходить за механізмом електрофільного заміщення (SE).
Нуклеофільне заміщення гідроксилу на галоген відбувається при дії галогенідів Фосфору та Сульфуру, а також галогеноводнів H–Hal (HI > HBr > HCl) на спирти за схемою:
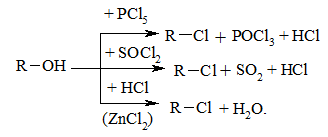
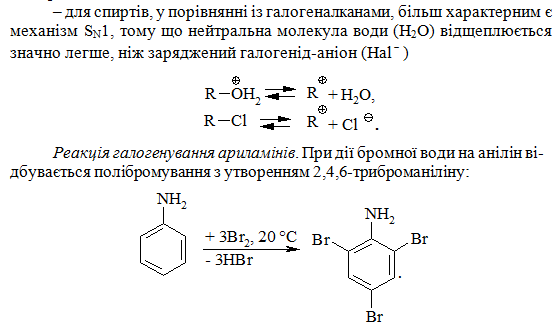
З кислотами НІ (50 мас %), HBr (50 мас %) та H2SO4 (96 мас %) реакція відбувається досить легко, з кислотою HCl значно важче в присутності каталітичних кількостей ZnCl2. З первинними спиртами реакція відбувається за механізмом SN2, а з третинними – SN1. Вторинні спирти реагують як за SN2, так і за SN1 механізмами.
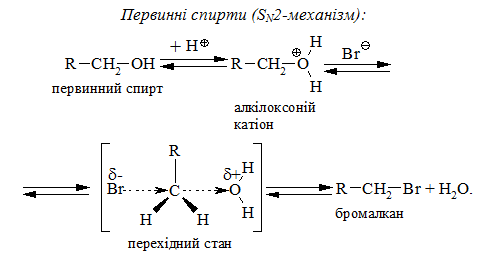
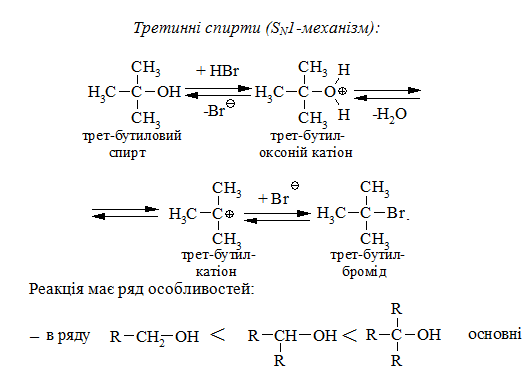
властивості спиртів посилюються, тому є закономірним зростання швид-кості реакції у тій же послідовності (з урахуванням протонування спиртів на першій стадії);
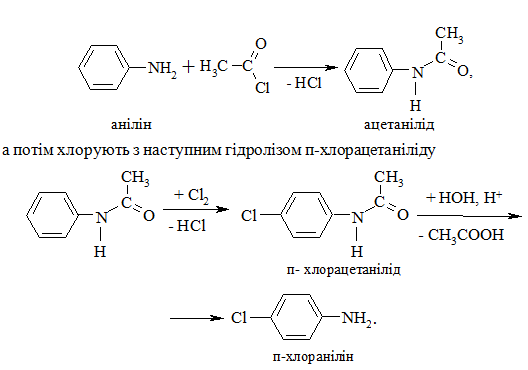
Аналогічно відбувається і йодування. При хлоруванні слід враховувати здатність аміногрупи до окиснення. Тому спочатку проводять захист аміногрупи ацилюванням:
5.3.1 Ацетилхлорид
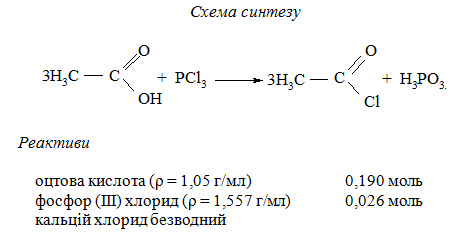
Методика синтезу. В колбу Вюрца об’ємом 200 мл вносять 0,19 моль оцтової кислоти і поміщають в баню з холодною водою. Колбу Вюрца з’єднують з краплинною лійкою похилим холодильником. Внутрішню трубку холодильника з’єднують з алонжем, боковий патрубок якого закритий хлоркальцієвою трубкою. Алонж з притертою пробкою опускають в конічну колбу. Ацетилхлорид легко розкладається вологою повітря, тому його не варто збирати у відкритому приймачі.
В колбу Вюрца при періодичному струшуванні за допомогою краплинної лійки поступово додають 0,026 моль фосфор(ІІІ) хлориду. Після додання розрахованої кількості фосфор (ІІІ) хлориду реакційну суміш нагрівають протягом 30 хв на водяній бані при температурі 40 – 50 ºС до тих пір, поки не припиниться бурхливе виділення хлороводню, а в рідині не утвориться дві фази. Верхній шар – ацетилхлорид, а фосфориста кислота – нижній.
Виділення продукту. Фази розділяють за допомогою ділильної лій-ки. Ацетилхлорид переносять у круглодонну колбу об'ємом 200 мл, поміщають на водяну баню, з'єднують з дефлегматором, термометром і похилим холодильником з алонжем. Алонж з'єднують з хлоркальцієвою трубкою і приймачем. В процесі перегонки відбирають фракцію, яка відганяється при температурі 50 – 53 ºС.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.10.
Характеристика кінцевого продукту. Ацетилхлорид – безбарвна рідина з різким запахом, димить на повітрі. Добре розчиняється в органічних розчинниках, Тпл = –112 °С, Ткип = 51,8 °С, d420 = 1,105, nd20 = 1,3898.
Токсичність. Подразнює шкіру, слизові оболонки дихальних шляхів та очей. ГДК р.з. = 0,1 мг/м3.
Використання. Як ацилюючий агент у виробництві фарб та лаків.
5.3.2 Бромбензол
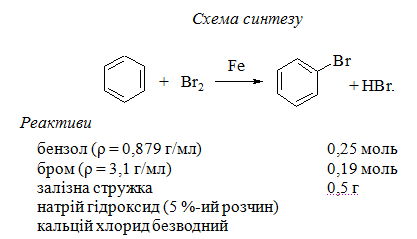
Методика синтезу. В круглодонну двогорлову колбу, оснащену краплинною лійкою і зворотним холодильником з газовідвідною трубкою, вносять 0,5 г залізної стружки та 0,25 моль попередньо висушеного бензолу, а потім частинами додають 0,19 моль брому з краплинної лійки. Після додання чергової порції брому вміст колби обережно перемішують. Залишок брому після початку виділення бромоводню доливають в колбу, не допускаючи бурхливого протікання реакції.
Для завершення реакції колбу протягом 30 хв нагрівають на теплій водяній бані (≈ 25 ºС), потім поступово підвищують температуру бані до 60 – 70 ºС. Нагрівання закінчують, коли над рідиною в колбі зникають бурі пари брому. Суміш виливають в колбу Вюрца об'ємом 50 мл і відганяють бромбензол з водяною парою. Змінивши приймач і зливши воду з холодильника, відганяють п-дибромбензол (побічний продукт реакції бромування). Дистилят з бромбензолом переносять в ділильну лійку, відділяють його від верхнього водного шару і сушать безводним кальцій хлоридом. З висушеного продукту відганяють з повітряним холодильником фракцію, яка кипить при температурі 152 – 158 ºС.
Виділення продукту. Отриманий продукт переносять в ділильну лійку, промивають водою, розведеним розчином натрій гідроксиду і знову водою, сушать кальцій хлоридом.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.11.
Характеристика кінцевого продукту. Бромбензол – важка прозора рідина. Нерозчинна у воді, добре розчинна в спирті, етері, хлороформі, бензолі, Тпл = 30,6 °С, Ткип = 156,2 °С, d420 = 0,1495, nd20 = 1,5602.
Токсичність. Уражає слизові оболонки дихальних шляхів, токсич-ний. ГДК – 3 мг/м3, ЛД50 = 2700 мг/кг.
Використання. В органічному синтезі (реакція Гриньяра, реакція Фіттіга).
5.3.3 1-Бромбутан
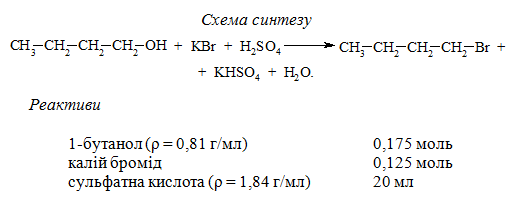
Методика синтезу. В конічну колбу на 250 мл вносять 10 мл дистильованої води і невеликими порціями (при перемішуванні та охолодженні колби під струменем води) додають 20 мл сульфатної кислоти. В тих же умовах (перемішування і охолодження!) до реакційної суміші додають 0,175 моль бутилового спирту.
В плоскодонну колбу на 250 мл вносять 0,125 моль калій броміду і невеликими порціями додають до нього приготовлену реакційну суміш (витяжна шафа!). До колби Вюрца приєднують похилий холодильник та алонж. Кінець алонжа занурюють в приймач з холодною водою. Реакційну суміш обережно нагрівають електронагрівачем, не допускаючи піноутворення. Реакцію вважають завершеною після повного розчинення калій броміду та закінчення стікання великих оліїстих крапель бутилброміду в приймач.
Виділення продукту. Бутилбромід відділяють від води через ділильну лійку, висушують безводним кальцій хлоридом та переганяють з колби Вюрца (Ткип = 101 – 102 oС).
Ця методика може використовуватись також для синтезу етил-, пропіл- та амілбромідів.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.12.

Характеристика кінцевого продукту. 1-бромбутан – безбарвна рідина з характерним запахом. Змішується з етиловим спиртом і діетиловим етером, добре розчиняється в ацетоні, Тпл = –111,9 °С, Ткип = 101,6 °С, d420 = 1,2588, nd20 = 1,4398.
Токсичність. ГДК р.з. = 0,3 мг/м3, ЛД50 = 4450 мг/кг.
Використання. Як напівпродукт в органічному синтезі.
Контрольні запитання
1. Яку реакцію називають реакцією галогенування? Наведіть її загальну схему.
2. Наведіть рівняння реакцїі отримання хлороцтової кислоти реакці-єю Хелля-Фольхарда-Зелінського.
3. Від яких чинників залежить швидкість реакції галогенування алканів галогенами?
4. Наведіть схему та механізм реакції бромування метану.
5. Як впливає будова алкану та природа галогену на можливість селек-тивного галогенування алканів?
6. Від яких чинників залежить швидкість галогенування алкенів галоге-нами?
7. Як змінюється реакційна здатність галогеноводнів в реакції гідрога-логенування алкенів? Наведіть схему реакції гідрогалогенування бутену-1 хлороводнем.
8. Наведіть схеми реакцій та зазначте вихід продуктів при взаємодії пропену-1 з йодоводнем без та за наявності пероксидів у системі.
9. За яким механізмом протікає реакція галогенування ароматичних вуглеводнів? Наведіть схему бромування бензолу. За яких умов вона протікає?
10. Наведіть схему та механізм реакції синтезу бромбутану з 1-бутано-лу.
5.4 Нітрування
Введення нітрогрупи –NO2 в органічну сполуку називається реакцією нітрування.
Найбільш поширеними нітруючими агентами є:
- суміш концентрованої нітратної та сульфатної кислот (нітруюча суміш);
- нітратна кислота різної концентрації в суміші з органічними розчинниками (HNO3 + ацетонітрил);
- тетраоксид нітрогену (N2O4 + ацетонітрил);
- алкілнітрити (Alk–NO2) або нітрити металів (NaNO2).
Найбільш поширені такі методи добування нітросполук:
- заміщення атома Гідрогену на нітрогрупу [41 – 50]:
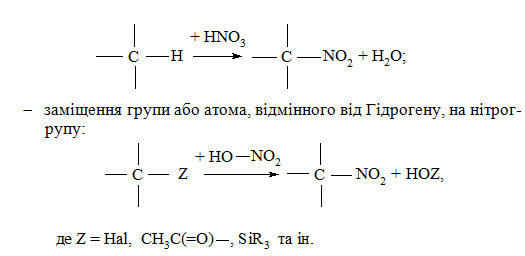
Наведемо деякі приклади реакції нітрування органічних сполук.
Нітрування алканів нітратною кислотою за звичайних умов не відбувається, а при нагріванні, як правило, іде їх окиснення. М. І. Коновалов (1889 р.) встановив, що при нагріванні (≈ 140 °С) під дією розведеної нітратної кислоти відбувається нітрування алканів за радикальним механізмом (SR) із задовільним виходом кінцевих нітроалканів:
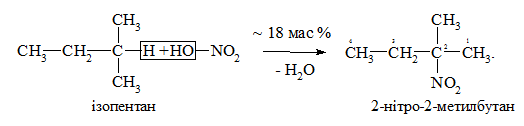
За таких умов забезпечується селективність нітрування (ізопентан нітру-ється переважно за третинним атомом Карбону). Парофазне нітрування (N2O4; Т ≥ 400 °C) не забезпечує селективності процесу нітрування.
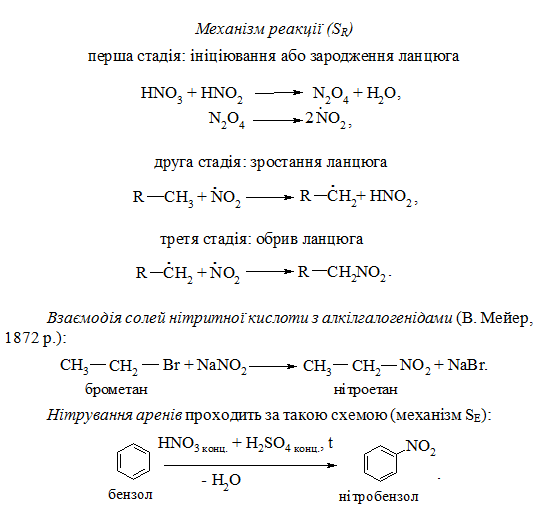
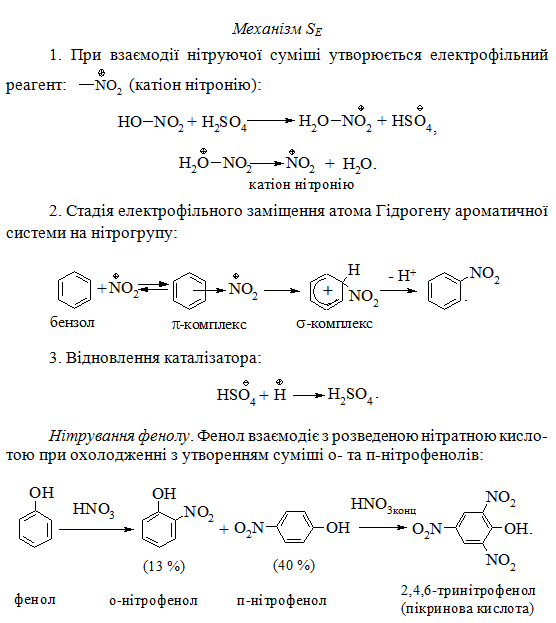
Застосування концентрованої нітратної кислоти веде до окиснення фенолу.
Нітрування карбонових кислот. В ароматичних карбонових кислотах карбоксильна група (carboxyl group) є потужним акцептором, який орієнтує нітрогрупу в метаположення ароматичного ядра в реакціях електрофільного заміщення (SЕ):
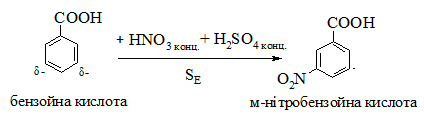
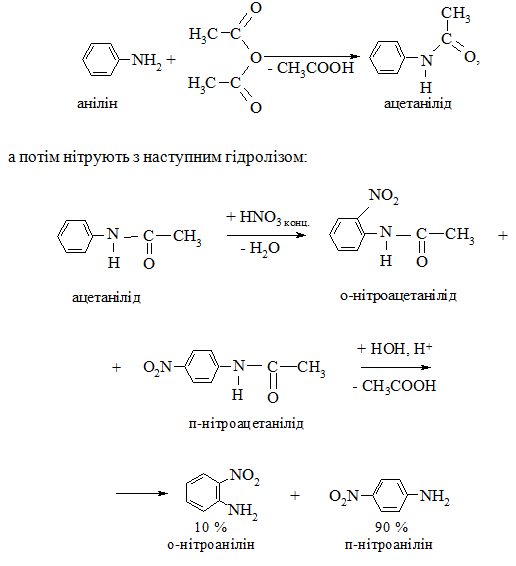
Нітрування амінів. Нітратна кислота є сильним окисником і для за-побігання окисненню аміногрупи, як і при хлоруванні, її захищають аци-люванням:
5.4.1 п-Нітроанілін
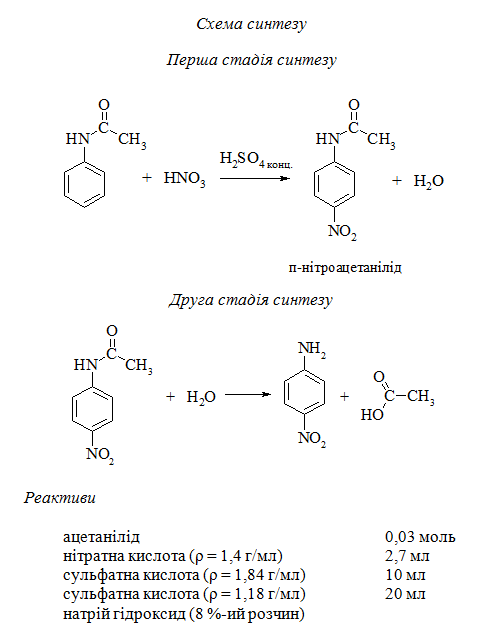
Синтез п-нітроаніліну проходить у дві стадії.
1. Добування п-нітроацетаніліду
Методика синтезу. Добре розтертий у ступці ацетанілід добавляють у колбу на 100 мл в кількості 0,03 моль, а потім розчиняють його у 7,5 мл концентрованої сульфатної кислоти (температура не повинна перевищувати 40 oС – заміряють температуру опущеним у колбу термометром). Розчин охолоджують до 5 oС в льодяній бані і повільно, при перемішуванні, додають до нього нітруючу суміш (суміш 2,5 мл концентрованої Н2SO4 та 2,7 мл концентрованої HNO3). Температура реакційної суміші при цьому не повинна перевищувати 15 oС.
Суміш залишають при кімнатній температурі на 45 хв, а потім виливають ії в колбу з 250 мл охолодженої льодом води. При цьому п-нітро-ацетанілід випадає у вигляді жовтого осаду, який відділяють вакуумним фільтруванням на лійці Бюхнера і промивають водою.
2. Добування п-нітроаніліну
Вологий п-нітроацетанілід шпателем переносять з паперового фільтра у плоскодонну колбу на 100 мл, додають 20 мл сульфатної кислоти (ρ = 1,18 г/мл) і нагрівають із зворотним холодильником до кипіння і повного розчинення п-нітроацетаніліду.
Виділення продукту. Гарячий розчин швидко фільтрують через складчастий фільтр. Фільтрат охолоджують до кімнатної температури і невеликими порціями додають до нього розчин 8 %-го натрій гідроксиду до нейтральної реакції (проба універсальним індикатором). При цьому п-нітро-анілін випадає у вигляді жовтого осаду.
Осад відфільтровують на вакуумній установці, перекристалізовують із води (Тпл = 147 – 148 oС).
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.13.

Характеристика кінцевого продукту. п-Нітроанілін – кристали жовтого кольору. Розчиняється у воді, етанолі, бензолі Тпл = 146,7 °С, Ткип = = 331 °С.
Токсичність. ГДКр.з. = 0,1 мг/м3.
Використання. В синтезі азобарвників, а також для отримання фенилендіамінів.
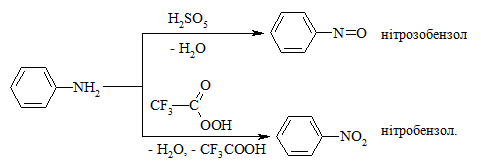
5.4.2 Нітробензол
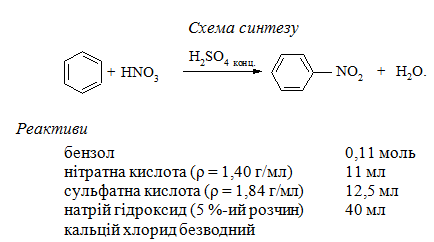
Методика синтезу. В тригорлову колбу об’ємом 100 мл вносять 11 мл нітратної кислоти і при охолодженні добавляють невеликим порціями 12,5 мл сульфатної кислоти. При необхідності суміш охолоджують до кімнатної температури (20 °С). Колбу з’єднують із зворотним холодильником, термометром та краплинною лійкою. Через краплинну лійку до нітруючої суміші, що вже знаходиться в колбі, порціями додають 0,11 моль бензолу, струшуючи кожного разу реакційну суміш (для цього закріплення колби в муфті повинно бути дещо ослаблене). Температура реакційної суміші при цьому не повинна перевищувати 50 °С (за необхідністю суміш охолоджують у заздалегідь приготовленій льодовій бані).
Після закінчення додавання бензолу реакційну суміш нагрівають на водяній бані до температури 60 °С та витримують її протягом 1 год.
Виділення продукту. Після закінчення реакції реакційну суміш охолоджують, переливають в ділильну лійку та залишають для відсто-ювання. Верхній шар (нітробензол) відділяють, промивають водою та двічі розчином натрій гідроксиду (2 х 20 мл) і знову водою (40 мл). Суміш переносять у ділильну лійку і відділяють нижній шар – нітробензол.
Промитий нітробензол вносять в суху колбу, приєднану до зворотного холодильника, додають безводний CaCl2 і нагрівають реакційну масу до її освітлення. При охолодженні реакційної маси нижній шар повинен затвердівати, утворюючи кристалогідрат CaCl2·6Н2О, якщо цього не відбувається, то в колбу додають ще CaCl2 та повторюють операцію до повного зневоднення нітробензолу.
Сухий нітробензол вносять в колбу Вюрца і переганяють його, відбираючи фракцію при 204 – 210 °С. Ні в якому разі нітробензол не переганяють досуха – це може призвести до вибуху. В колбі повинно залишитись 10 – 15 % початкового об’єму.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.14.
Характеристика кінцевого продукту. Нітробензол чистий – безбарвна рідина, технічний – рідина світло-жовтого кольору. Легко розчинний в етанолі, етері, бензолі, нерозчинний у воді, Тпл = 5,76 °С, Ткип = 210,8 °С, d420 = 1,208, nd20 = 1,5526.
Токсичність. Дуже отруйна речовина, окиснює гемоглобін до метгемоглобіну, порушує діяльність центральної нервової системи, викликає захворювання печінки. ГДКр.з.= 3 мг/м3.
Використання. Для отримання аніліну, а також бензидину, м-ди-нітробензолу, м-нітрохлорбензолу, м-нітробензолсульфокислоти; в виробництві барвників як розчинник при очищенні сирої нафти, а також використовується як м'який окисник.
5.4.3 м-Нітробензойна кислота
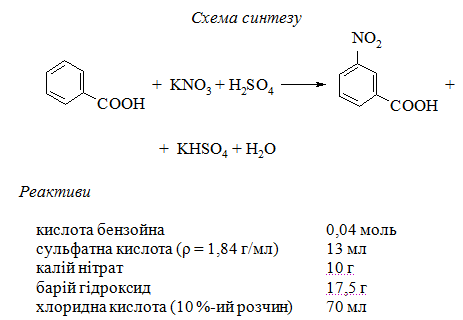
Методика синтезу. В двугорлову колбу, оснащену мішалкою і термометром, вносять 13 мл концентрованої сульфатної кислоти і нагрівають на водяній бані до 70 ºС. Баню відставляють і при постійному перемішуванні додають суміш 0,04 моль бензойної кислоти і 10 г калій нітрату. Температура реакційної суміші не повинна перевищувати 80 ºС.
Потім реакційну суміш нагрівають на водяній бані (≈ 85 – 90 ºС) до тих пір, поки на поверхні реакційної маси не утвориться маслянистий шар м-нітробензойної кислоти. Розчин охолоджують і виливають в холодну воду. Осад м-нітробензойної кислоти, що утворився, фільтрують та промивають спочатку холодною, а потім 2 – 3 рази гарячою водою.
Виділення продукту. Осад переносять в колбу для перегонки з водяною парою і відганяють залишок бензойної кислоти, який не прореагував. м-Нітробензойну кислоту виділяють у вигляді її барієвої солі. Осад кислоти розчиняють в 20-кратній (за масою) кількості води і обробляють гарячим розчином барій гідроксиду (17,5 г в 50 мл води) до слабко лужної реакції. Потім додають 200 мл води і кип'ятять суміш до повного розчинення осаду. Розчин фільтрують через лійку для гарячого фільтрування, фільтрат охолоджують і продукт відфільтровують.
Для виділення вільної кислоти її барієву сіль кип'ятять в 70 мл 10 %-го розчину хлоридної кислоти. Після охолодження осад м-нітробензойної кислоти відфільтровують і промивають холодною водою.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.15.
Характеристика кінцевого продукту. м-Нітробензойна кислота – безбарвна кристалічна речовина. Розчинна в етанолі, етері, важкорозчинна в бензолі і холодній воді, Тпл = 141 °С, d420 = 1,494.
Використання. Для виробництва бензидин-3,3-дикарбонової кислоти, для осадження Th, Ce, Zr, Hg, Hf для мікрокристалічного виявлення алкалоїдів, у виробництві лікарських препаратів.
Контрольні запитання
1. Яку реакцію називають реакцією нітрування? Назвіть найбільш по-ширені нітруючі агенти.
2. Назвіть основні методи добування нітросполук. Наведіть схеми реакцій.
3. Чи використовують концентровану нітратну кислоту при прямому нітруванні алканів? За яких умов його здійснюють?
4. Наведіть схему та механізм реакції нітрування етану розбавленою нітратною кислотою.
5. За яких умов при нітруванні бутану отримують переважно 2-нітро-бутан?
6. Наведіть схему реакції отримання 1-нітропропану при використанні натрій нітриту як нітруючого агенту.
7. Наведіть схему та механізм реакції нітрування бензолу.
8. Які продукти утворюються при нітруванні толуолу? Наведіть схему реакції.
9. Які ізомери (о-, м-, п-) переважно отримують при нітруванні ароматичних фенолів? Ароматичних карбонових кислот?
10. Чому синтез п-нітроаніліну з аніліну здійснюють у декілька стадій?
5.5 Сульфування
Сульфування (sulfonation) – це введення сульфогрупи –SO3H в молекулу органічної сполуки за допомогою таких найбільш поширених способів.

В промисловості широко використовується сульфоокиснення або сульфохлорування (sulfochlorination):

Електрофільне приєднання сульфатної кислоти до алкенів прохо-дить за правилом Марковнікова з утворенням кислих алкілестерів сульфатної кислоти, гідроліз яких приводить до утворення відповідних спиртів:
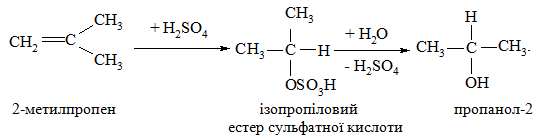
Таким методом в промисловості отримують етанол та пропанол-2.
Сульфування аренів проходить при дії концентрованої сульфатної кислоти за схемою:
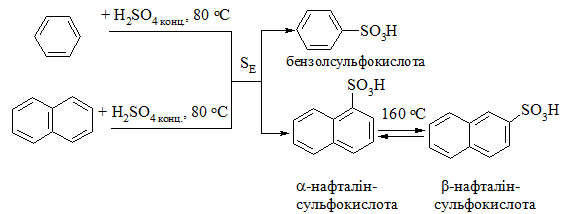
Необхідно відмітити, що при нагріванні до 160 °С α-нафталінсульфокис-лота переходить в β-ізомер.
Феноли легко сульфуються концентрованою сульфатною кислотою навіть за кімнатної температури:
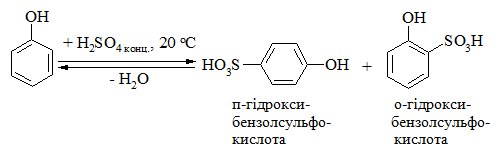
з наступною селективною ізомеризацією утвореної о-гідрокси-бензолсульфокислоти при нагріванні до 100 °С до п-ізомера. Так при температурі 20 °С утворюється 90 % о-ізомеру і 10 % п-ізомеру, а при 100 °С – 10 % о-ізомеру і 90 % п-ізомеру.
5.5.1 Натрій п-толуолсульфонат
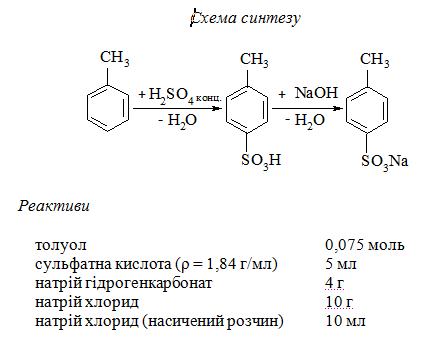
Методика синтезу. В круглодонну колбу на 200 мл вносять 0,075 моль толуолу і при перемішуванні невеликими порціями додають 5 мл сульфатної кислоти. Суміш перемішують впродовж 10 хв. Колбу з’єднують із зворотним холодильником і, струшуючи її через кожні 5 хв, нагрівають на електроплитці. Нагрівання продовжують протягом 30 – 40 хв при слабкому кипінні до зникнення шару толуолу.
Виділення продукту. Реакційну суміш охолоджують і теплою вили-вають в конічну колбу, заповнену 30 мл води. Кислий розчин частково нейтралізують, при доданні невеликими порціями твердого натрій гідрогенкарбонату (обережно – спінення!). Потім додають твердий натрій хлорид та нагрівають суміш до повного розчинення солі.
Гарячий розчин фільтрують через лійку для гарячого фільтруван-ня. З охолодженого фільтрату кристалізується натрій п-толуолсульфонат. Кристалічний осад відділяють вакуумним фільтруванням на лійці Бюхнера, промивають 10 мл насиченого розчину натрій хлориду, висушують і зважують.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.16.
Характеристика кінцевого продукту. Натрій п-толуолсульфонат – безбарвна кристалічна речовина. Добре розчинна у воді, розчинна в етанолі, етері, гігроскопічна, на повітрі розпливається Тпл = 107°С, Ткип = 140 °С.
Використання. Для виробництва п-крезолу, азобарвників, дезинфекційних відбілювальних речовин, хлораміну-Т.
5.5.2 Сульфанілова кислота
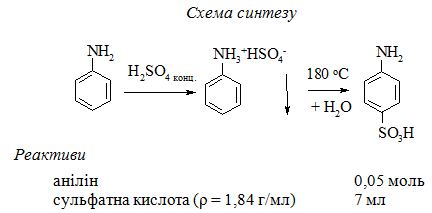
Методика синтезу. В плоскодонну колбу з вузькою горловиною на 100 мл заливають 7 мл концентрованої сульфатної кислоти і при обережному перемішуванні невеликими порціями додають 0,05 моль аніліну (так, щоб він не потрапив на горловину колби). Колбу закріплюють над електроплиткою і опускають в неї термометр таким чином, щоб ртутна кулька термометра була занурена у реакційну суміш (колбу пробкою не закривати!). Нагрівання продовжують протягом години при температурі 170 oС.
Виділення продукту. Гарячу реакційну суміш обережно виливають в колбу з 70 мл охолодженої води при постійному перемішуванні. Білий кристалічний осад сульфанілової кислоти відфільтровують під вакуумом на лійці Бюхнера, промивають на фільтрі холодною водою, перекристалізовують з води і висушують на повітрі. При цьому виділяється кристалогідрат сульфанілової кислоти. При висушуванні у сушильній шафі (100 – 105 oС) утворюється безводна сульфанілова кислота.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.17.
Характеристика кінцевого продукту. Сульфанілова кислота – без-барвна кристалічна речовина. Важкорозчинна в більшості органічних розчинників і холодній воді. При температурі 100 °С втрачає кристалізаційну воду, при нагріванні до 280 °С розкладається без плавлення.
Використання. Для лабораторного визначення нітритів, Осмію, Рутенію, Церію (IV).
Контрольні запитання
1. Яку реакцію називають реакцією сульфування? Назвіть найбільш поширені сульфівні реагенти.
2. Назвіть основні способи сульфування. Наведіть схеми реакцій.
3. Прямим сульфуванням яких алканів (бутану чи метилбутану) концентрованою сульфатною кислотою отримують алкансульфокислоту? Наведіть відповідне рівняння реакції.
4. Наведіть схему реакції отримання сульфокислоти з н-пропану.
5. За яким механізмом протікає реакція сульфоокиснення?
6. Наведіть схему сульфохлорування етану з наступним гідролізом отриманого продукту.
7. Який сульфівний реагент використовують при отриманні етанолу? Наведіть відповідні рівняння реакцій.
8. Наведіть рівняння реакцій взаємодії 1-пропену з сульфатною кисло-тою та гідролізу отриманого продукту.
9. Наведіть схему реакції сульфування нафталіну. Який сульфівний реагент при цьому використовують?
10. За яких умов при сульфуванні фенолу отримують переважно п-гідроксибензолсульфокислоту? Наведіть схему реакції.
5.6 Діазотування та азосполучення
Діазотування (diazotization) – це перетворення ароматичних амінів в солі арилдіазонію під дією суміші нітритної та мінеральної кислот (реакція П. Гріса, 1858 р.) [41, 59 – 61]:

Деякі особливості проведення реакції
1. Нітритна кислота – HNO2 та солі діазонію ArN2+Cl- нестійкі і розкладаються уже за кімнатної температури, тому реакцію необхідно проводити при охолодженні до 0 – 5 °С.
2. Механізм реакції, відповідно до результатів кінетичних дослі-джень Інгольда, іонний, тому чим більшою є нуклеофільність атома Нітрогену, тим більшою є швидкість реакції.
3. В реакції необхідно використовувати 2,5 – 3,0 мольний надлишок хлоридної кислоти: 1 моль витрачається на утворення нітритної кислоти, ще 1 моль – на утворення солі діазонію, а надлишок (0,5 – 1,0 моль) для створення кислого середовища.
Інші азосполуки добувають на основі солей діазонію.
Цей метод синтезу широко застосовується у промисловості для синтезу барвників та різних органічних сполук.
Азосполученння – це отримання азосполук (Ar–N=N–Ar') із солей діазонію та ароматичних амінів, фенолів та етерів за схемою [10 – 12]:
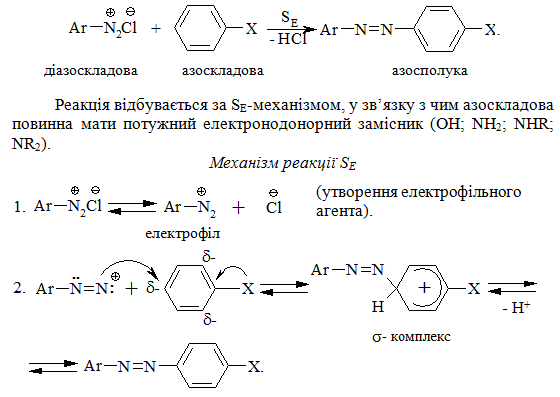
Деякі особливості проведення реакції
1. Катіон арилдіазонію – електрофільний реагент, тому введення електроноакцепторних замісників в ароматичне ядро підвищує як їх тер-модинамічну стійкість, так і його реакційну здатність за рахунок збіль-шення позитивного заряду на кінцевому атомі Нітрогену. Стійкість солей арилдіазонію зменшується в ряду:
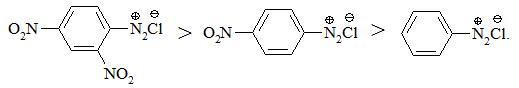
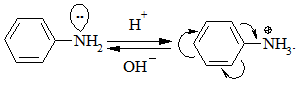
2. З ароматичними амінами реакція відбувається у слабокислому середовищі, в якому аміногрупа переходить в амонійну форму, яка дезактивує ароматичне ядро через зменшення його нуклеофільності:
При застосуванні фенолів як азоскладових реакцію проводять у слаболужному середовищі, в якому вони перетворюються у більш активний фенолят-аніон:
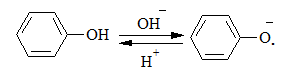
3. Враховуючи незначну термодинамічну стійкість солей арилдіазонію, реакцію, як правило, проводять при температурі 0 – 5 °С. При добуванні більш стійких солей діазонію реакцію можна проводити при кімнатній температурі.
4. Якщо у пара-положенні азоскладової знаходиться замісник, то реакція азосполучення відбувається у незаміщене ортоположення.
Азосполуки широко використовуються як барвники. При цьому їх забарвлення та його інтенсивність залежить від будови. Молекули азосполук містять хромофорні групи:
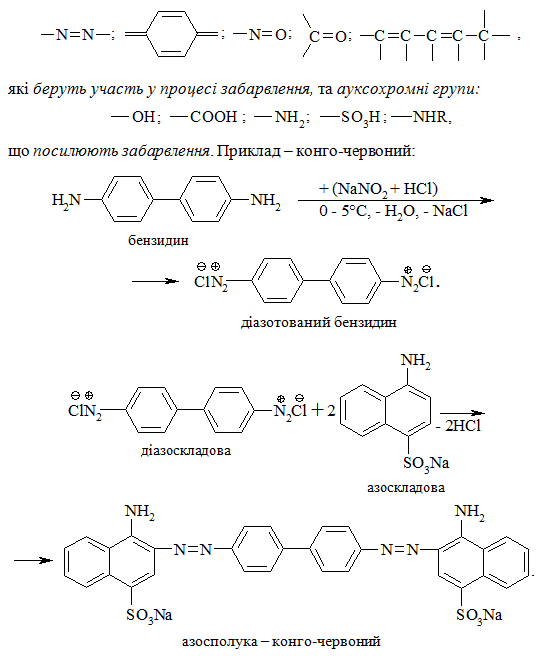
Добре знайомий аналітичний індикатор метиловий оранжевий, що змінює своє забарвлення під впливом рН середовища, також є азобарвником:
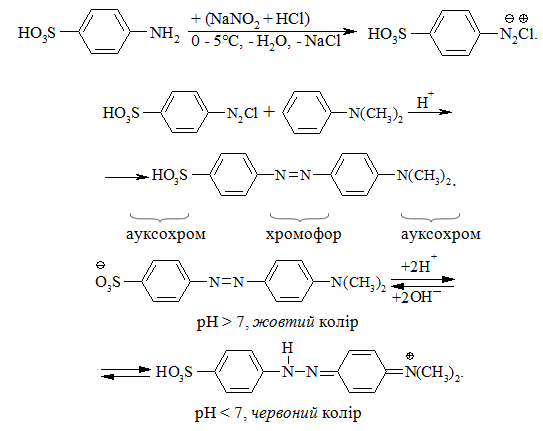
Такі сполуки широко використовують як кислотно-основні індикатори у аналітичній практиці.
5.6.1 β-Нафтолазо-п-нітробензол
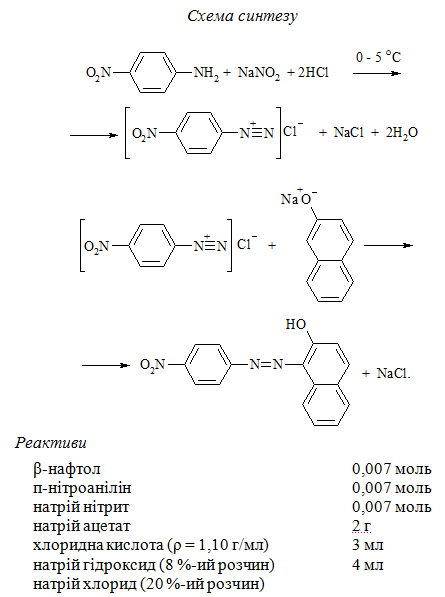
Методика синтезу
Діазотування. Наважку п-нітроаніліну засипають в конічну колбу, розчиняють в 10 мл гарячої води, додають 1,5 мл хлоридної кислоти, охолоджують до 0 oС в льодяній бані і додають ще 1,5 мл хлоридної кислоти та 10 мл води. Розчин п-нітроаніліну діазотують, додаючи невеликими порціями при перемішуванні розчин натрій нітриту 0,007 моль в 4 мл води. Закінчення процесу визначають за йод-крохмальним індикатором. Реакційну суміш залишають при 0 oС на 30 хв, а потім додають розчин натрій ацетату, який містить 2 г солі в 7 мл води. Отримують розчин солі діазонію (розчин І).
Азосполучення. Наважку β-нафтолу 0,007 моль засипають в конічну колбу, додають 4 мл 8 %-го розчину натрій гідроксиду та 65 мл гарячої води. Розчин охолоджують і при перемішуванні додають до нього раніше отриману сіль діазонію (розчин І). Через 30 хв барвник відфільтровують, промивають 20 %-ним розчином натрій хлориду, водою і висушують на повітрі.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.18.
Характеристика кінцевого продукту. β-Нітроаніліновий червоний – кристалічна речовина яскраво-червоного кольору. Нерозчинна у воді.
Використання. Як азобарвник в промисловості.
Контрольні запитання
1. Яку реакцію називають реакцією діазотування? Наведіть приклади та схеми реакцій.
2. Наведіть механізм реакції діазотування.
3. Чому діазотування проводять при охолодженні реакційної суміші?
4. Яку реакцію називають реакцією азосполучення? Наведіть схему та приклади реакцій.
5. Наведіть механізм реакції азосполучення.
6. В якому середовищі і чому проводять реакцію азосполучення при використанні як азоскладової амінів, фенолів?
7. Як впливає будова вихідного ароматичного аміну на стійкість солей діазонію?
8. Наявність яких груп у складі азосполук зумовлює їх забарвлення?
9. Наведіть схему синтезу метилового оранжевого.
10. Наведіть взаємоперетворення метилового оранжевого в залежності від рН розчину.
5.7 Окиснення
Введення атома Оксигену в молекулу органічної сполуки або вилу-чення атома Гідрогену при його заміщенні на Оксиген називається реакцією окиснення.
В більш загальному вигляді реакцію окиснення можна розглядати як процес зміни ступеня окиснення атома Карбону в органічній молекулі з урахуванням електронегативності хімічних елементів, що входять до її складу. Наявність в органічних молекулах ковалентного зв'язку викликає необхідність введення терміну "умовного ступеня окиснення" хімічних елементів, що входять до їх складу.
При цьому ступінь окиснення найбільш поширених елементів може змінюватися в межах:

Але окисно-відновні реакції органічних сполук можуть бути пов'язані не лише зі зміною кількості атомів Оксигену у їх складі. Тому процес окиснення органічних сполук можна розглядати більш широко в реакціях заміщення, приєднання та елімінування [62 – 64].
Заміщення Гідрогену або інших атомів на Оксиген може проходити за такими схемами:
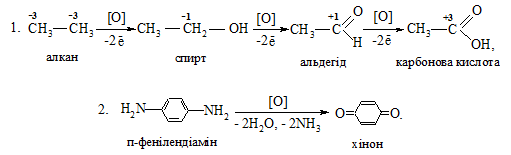
Приєднання до подвійного Карбон-Карбонового зв'язку та приєднання атома Оксигену до атомів Карбону змінної валентності:
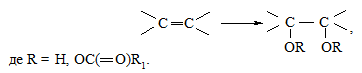
Елімінування (відщеплення) Гідрогену з утворенням кратних зв'язків між атомами:
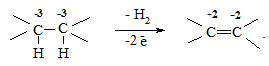
Найбільш поширеними окисниками є:
- кисень повітря (О2 + Kat; Kat: Al2O3, Cr2O3, MnO2, солі купруму, плюмбуму, мангану та ін.);
- калій перманганат (KMnO4 в різних середовищах);
- натрій і калій дихромат (Na2Cr2O7, K2Cr2O7 широко використовують для окиснення первинних і вторинних спиртів до альдегідів та кетонів, відповідно);
- хромовий ангідрид (CrO3 в розчині оцтової кислоти);
- нітратна кислота (HNO3 (10 – 25 мас %) або HNO3 (65 – 95 мас %) на холоді або при нагріванні);
- гідроген пероксид (H2O2 в різних середовищах).
Розглянемо деякі конкретні приклади реакції окиснення органічних сполук.
Окиснення алканів може бути повним (горіння) та неповним (утво-рення спиртів, альдегідів, кетонів та карбонових кислот).
Алкани, як паливо, горять із виділенням великої кількості теплової енергії (повне окиснення):
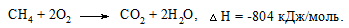
У випадку неповного окиснення (окисники: О2 повітря, KMnO4, K2Cr2O7; температура: ≈ 150 – 200 °С; каталізатор: MnO2, Mn(СН3СОО)2) утворюється суміш оксигеновмісних вуглеводнів: спиртів, альдегідів, кетонів та карбонових кислот. Механізм реакції радикально-ланцюговий (SR):
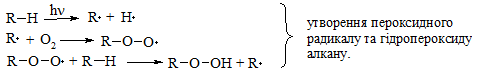
Вторинні гідропероксиди алканів утворюють при окисненні дещо інші продукти реакції:
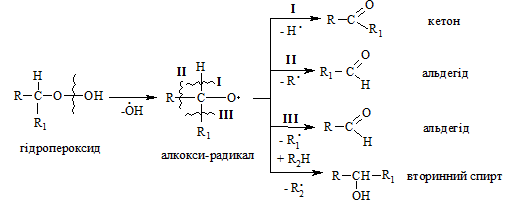
У випадку третинних гідропероксидів алканів реакція окиснення проходить за такою схемою:
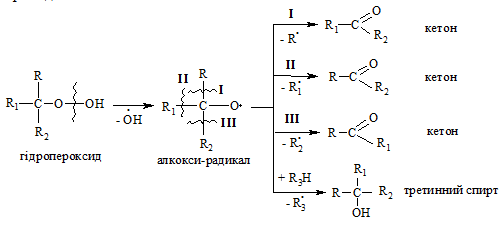
Особливості процесу окиснення:
- гомолітичний розрив зв’язку С–С завжди проходить в -поло-женні відносно атома Оксигену алкокси-радикалу (на схемах позначення І, ІІ, ІІІ);
- спирти та альдегіди окиснюються далі до карбонових кислот або кетонів;
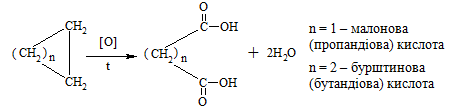
- окиснення циклоалканів, як і у випадку парафінів, проходить в „жорстких” умовах з утворенням циклічних кетонів, спиртів, а також ди-карбонових кислот при розриві циклу та збереженні загальної кількості атомів Карбону у вуглеводневому ланцюзі:
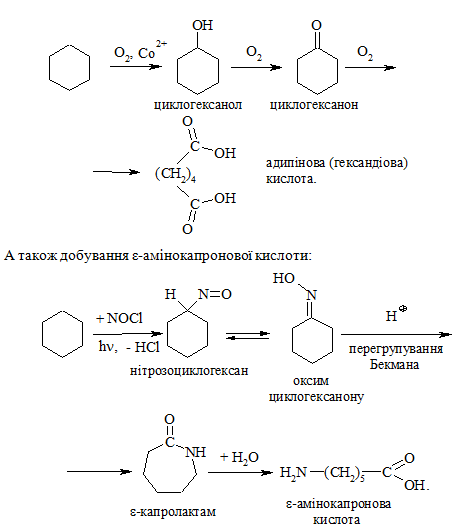
Промислове значення має добування адипінової кислоти окисненням циклогексану в присутності каталізатора (нафтенату кобальту або мангану):
Окиснення алкенів із розривом подвійного зв’язку. Реакція відбува-ється в жорстких умовах із використанням сильних неорганічних окисників (калій перманганату або дихромату в середовищі концентрованої сульфатної кислоти при нагріванні):
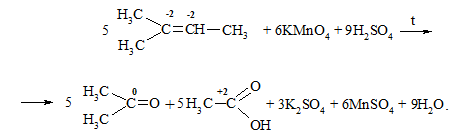
Електронний баланс реакції:

Дизаміщені похідні алкінів теж окиснюються з розривом потрійного зв’язку:
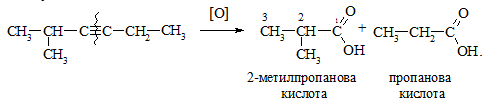
Цю реакцію алкінів можна використовувати для визначення будови алкі-нів, аналізуючи карбонові кислоти, які при цьому утворюються.
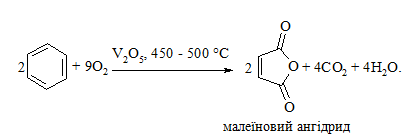
Окиснення ароматичних сполук. Арени за звичайних умов стійкі до дії сильних окисників (KMnO4, K2Cr2O7, CrO3). Реакція відбувається в “жорстких” умовах із використанням V2O5 як каталізатора:
Гомологи бензолу, що мають бокові вуглеводневі ланцюги, окиснюються значно легше:
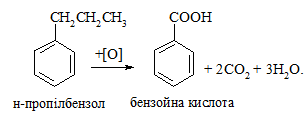
Особливості окиснення гомологів бензолу:
- незалежно від довжини бічного ланцюга він окиснюється до кін-цевої карбоксильної групи;
- при наявності кількох бокових ланцюгів, кожний наступний окиснюється у більш “жорстких” умовах, ніж попередній.
Арени взаємодіють із озоном за схемою:
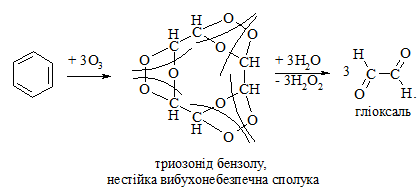
Окиснення спиртів проводять, як правило, сильними окисниками: (K2Cr2O7 + H2SO4), CrO3, (KMnO4 + H2SO4), киснем повітря (у промисловості). При цьому окиснюється атом Карбону, який безпосередньо зв’язаний із гідроксильною групою, тому в залежності від будови спирту (первинний, вторинний або третинний), утворюються різні продукти реакції.
Первинні спирти окиснюються в залежності від умов реакції за схемою:
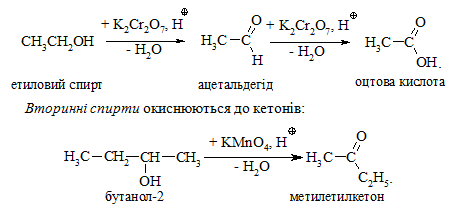
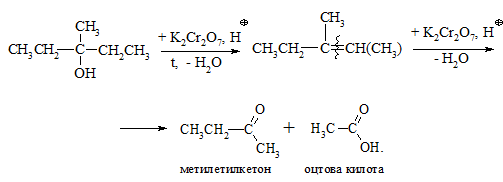
Третинні спирти окиснюються в жорстких умовах спочатку з утворенням алкенів, а потім, з розривом подвійного зв’язку, до кетонів та карбонових кислот з меншим, ніж у молекулі вихідного спирту, числом атомів Карбону:
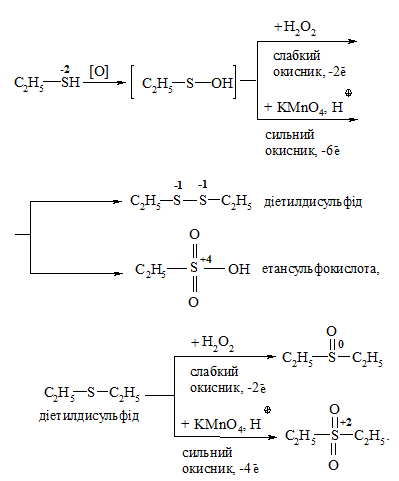
Тіоспирти (меркаптани та тіоетери (сульфіди)) окиснюються досить легко за атомом Сульфуру і, залежно від умов проведення реакції, утворюють різні сполуки:
Аміни окиснюються різними окисниками. Продукти реакцій окис-нення залежать як від будови аміну, так і від природи окисника:
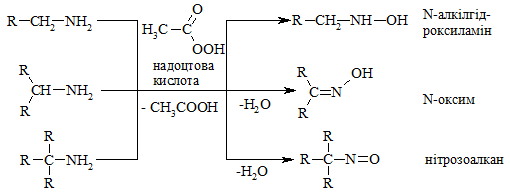
Первинні ароматичні аміни окиснюються кислотою Каро – H2SO5 до нітрозосполук, а трифторнадоцтовою кислотою – до нітропохідних аренів:
5.7.1 Ацетон
Схема синтезу
Методика синтезу. В двугорлову колбу, оснащену краплинною лійкою і зворотним холодильником, поміщають 0,26 моль ізопропілового спирту. В конічній колбі готують хромову суміш: розчиняють 15 г натрій дихромату в 60 мл води і змішують отриманий розчин з 18 мл концентрованої сульфатної кислоти (обережно додають кислоту в водний розчин натрію дихромату). Отриману хромову суміш з краплинної лійки невеликими порціями (по 1 – 2 мл) доливають в колбу. Окиснення супроводжується сильним нагріванням реакційної маси. Після додання хромової суміші колбу нагрівають на водяній бані впродовж 10 хв.
Виділення продукту. Після охолодження реакційну суміш перено-сять в колбу Вюрца з холодильником Лібіха і відганяють ацетон на водяній бані, збираючи фракцію з Ткип = 55 – 58 ºС.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.19.

Характеристика кінцевого продукту. Ацетон – безбарвна прозора рідина з характерним запахом. Змішується з водою в усіх співвідношеннях Тпл = –95,35 °С, Ткип = 56,24 °С, d420 = 0,7908, nd20 = 1,3558.
Токсичність. Наркотична речовина, яка поступово вражає всю центральну нервову систему. ГДКр.з. = 200 мг/м3, ЛД50 = 3800 мг/кг.
Використання. Як розчинник багатьох органічних речовин, в першу чергу ацетилену, нітро- та ацетилцелюлози; є сировиною для синтезу великої кількості сполук.
5.7.2 Антрахінон
Схема синтезу
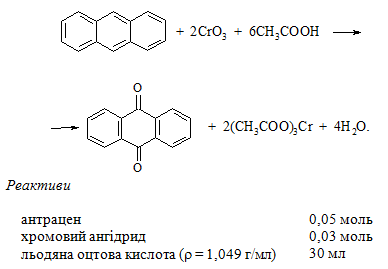
Методика синтезу
Приготування розчину хромового ангідриду. Розтертий у ступці хромовий ангідрид зважують і розчиняють в 10 мл льодяної оцтової кислоти. Потім додають невелику кількість води до повного розчинення ангідриду та переносять розчин у краплинну лійку.
Окиснення антрацену до антрахінону. Плоскодонну колбу на 200 мл через насадку з’єднують із зворотним холодильником та краплинною лійкою. В колбу завантажують суміш антрацену з 20 мл оцтової кислоти і нагрівають її до кипіння. Далі з краплинної лійки невеликими порціями протягом 20 хв додають приготовлений розчин хромового ангідриду.
Виділення продукту. Суміш нагрівають ще 20 хв, а потім обережно її виливають в конічну колбу з 50 мл холодної води. Антрахінон утворює осад, який відфільтровують на лійці Бюхнера з допомогою вакуумного фільтрування.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.20.
Характеристика кінцевого продукту. Антрахінон – світло-жовті ромбічні кристали. Розчиняється в нітробензолі, аніліні і гарячому толуолі; малорозчинний в спирті; в H2SO4 конц. розчиняється з утворенням жовтого забарвлення, Тпл = 286 °С, Ткип = 379 °С, d420 = 1,348.
Токсичність. Викликає екзему та бронхіальну астму. ГДКр.з. = = 5 мг/м3, ЛД50 = 3500 мг/кг
Використання. У виробництві антрахінонових барвників, антиоксидантів, інгібіторів (inhibitors) полімеризації (polymerization), в паперовій промисловості для підвищення міцності целюлозних волокон.
5.7.3 Бензойна кислота
Схема синтезу
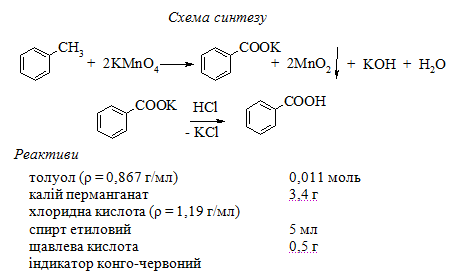
Методика синтезу. В круглодонну колбу, оснащену зворотним кульковим холодильником, вносять 0,011 моль толуолу, 3,4 г тонкоподрібненого калій перманганату і 75 мл води. Колбу нагрівають на водяній бані протягом 2 годин. Якщо розчин після нагрівання зберігає рожеве забарвлення, то до гарячої, але не киплячої реакційної суміші, через холодильник обережно додають 3 – 5 мл етилового спирту або 0,5 г щавлевої кислоти.
Після закінчення реакції безбарвний розчин охолоджують, осаджений манган(ІV) оксид відфільтровують і промивають на фільтрі киплячою водою (2 порціями по 15 мл). Промивні води об'єднують з фільтратом і упарюють на водяній бані до об'єму 10 – 15 мл. Якщо знову випадає осад манган(ІV) оксиду, його відфільтровують.
Виділення продукту. До упареного розчину калій бензоату краплинами додають концентровану хлоридну кислоту до кислої реакції за конго-червоним. Виділений осад бензойної кислоти відфільтровують, промивають невеликою кількістю холодної води (10 мл) і висушують на повітрі.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.21.
Характеристика кінцевого продукту. Бензойна кислота – безбарвна кристалічна речовина зі слабким запахом. Погано розчинна в холодній воді, етері, добре – в спирті, оцтовій кислоті, піридині, Тпл = 122 °С, Ткип = 249 °С, d420 = 1,2659, nd20 = 1,5397.
Токсичність. Проявляє слабку наркотичну дію, але меншу ніж у бензолу; діє на центральну нервову систему та органи кровотворення. ЛД50 = 1700 мг/кг.
Використання. В синтезі барвників різних класів та фармацевтичних препаратів; сировина для отримання капролактаму; добавка до бензинів для підвищення октанового числа (octane number).
Контрольні запитання
1. Яку реакцію називають реакцією окиснення? Як змінюється ступінь окиснення найбільш поширених елементів в молекулах органічних ре-човин?
2. При протіканні яких реакцій відбувається окиснення органічних речовин? Як змінюється ступінь окиснення атома Карбону в ряду: насичений вуглеводень – спирт – альдегід – карбонова кислота?
3. Назвіть речовини, які використовуються як окисники в процесах окиснення органічних речовин.
4. Наведіть схему та механізм реакції окиснення етану до етаналю.
5. Наведіть схему та механізм реакції окиснення пропану до диметил-кетону.
6. Наведіть схему та механізм реакції окиснення метилпропану до 2-метил-2-пропанолу.
7. Наведіть рівняння реакції окиснення пропену калій перманганатом. Поставте коефіцієнти методом електронного балансу. Які продукти утворюються при озонолізі пропену?
8. За яких умов окиснюються бензол та його гомологи? З яких вихідних речовин можна отримати бензойну кислоту: бензол, толуол, етилбензол, п-ксилол? Наведіть відповідні рівняння реакцій.
9. Які продукти утворюються при окисненні 1-пропанолу; 2-пропанолу; 2-метил-2-пропанолу? Наведіть відповідні рівняння реакцій.
10. Як впливає природа окисника та будова аміну на продукти їх окиснення?
5.8 Відновлення
Приєднання Гідрогену до кратних зв’язків молекули органічної спо-луки або вилучення атома Оксигену, його заміщення на Гідроген називається реакцією відновлення. Відновлення – це складова загальної окисно-відновної реакції, тому необхідно вивчити загальні положення стосовно таких реакцій, які викладено в підрозділі 5.7 Окиснення. Процеси відновлення органічних сполук проходять в результаті реакцій заміщення, приєднання та елімінування [65 – 69].
Заміщення Оксигену або інших атомів на атом Гідрогену:
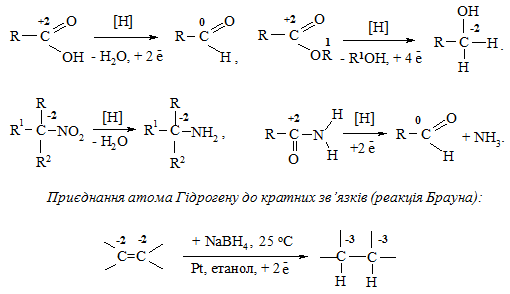
або одночасне приєднання Гідрогену та заміщення атома Оксигену у випадку α, β-епоксикетонів (реакція Уортона):
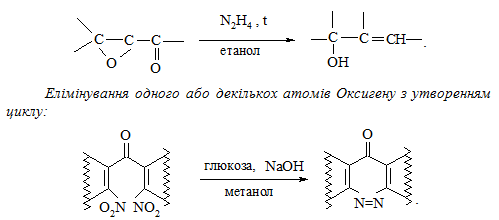
Розглянемо деякі конкретні приклади реакції відновлення. Гідрування (гідрогенізація) алкенів у більшості випадків відбувається при дії каталізаторів платинової групи (Pt, Pd, Ni). В залежності від умов проведення реакція може відбуватись за механізмом радикального (AR) або електрофільного (АЕ) приєднання:
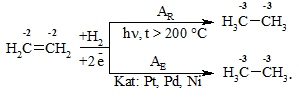
Реакція гідрування алкінів (приєднання водню) проходить у присутності каталізаторів стереоселективно з утворенням продуктів цис- та транс-приєднання:

Реакція гідрування для аренів малохарактерна і потребує більш жорстких умов, ніж у випадку алкенів та алкінів.
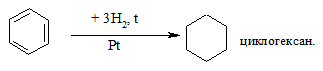
Реакція відбувається до повного насичення ароматичного ядра та утворення циклогексану, тому зупинити її на стадії часткового гідрування неможливо.
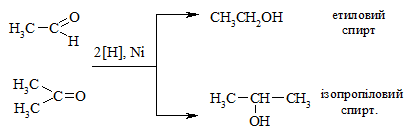
Відновлення альдегідів та кетонів. Каталітичне гідрування можна проводити за різних умов, отримуючи первинні або вторинні спирти:
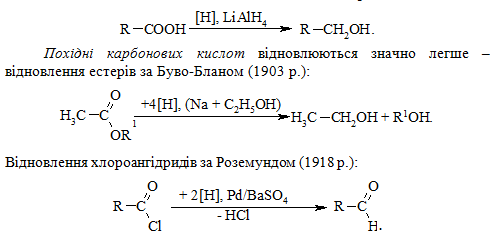
Реакції відновлення аліфатичних та ароматичних карбонових кислот відбуваються важко. Але під дією літій тетрагідроалюмінату LiAlH4 вони досить легко відновлюються до відповідних спиртів:
Реакція відновлення нітроаренів залежить від рН середовища та природи відновника. В нейтральному та кислому середовищі (відновники Fe + HCl; Sn + HCl; Sn) реакція відбувається за схемою:
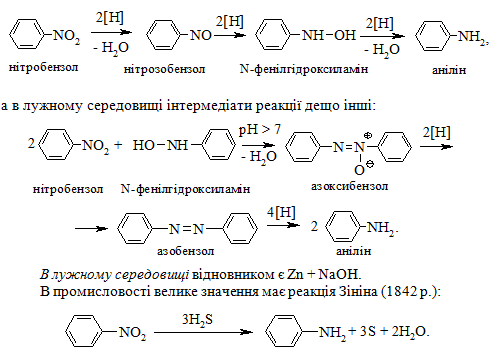
5.8.1 Анілін
Схема синтезу
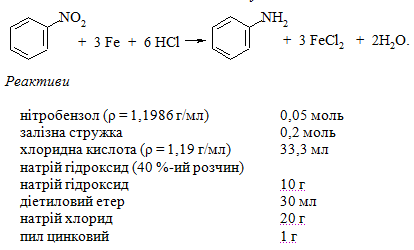
Методика синтезу. В круглодонній колбі із зворотним повітряним холодильником змішують 0,05 моль нітробензолу і 0,2 моль залізної стружки. Далі при постійному струшуванні невеликими порціями додають 33,3 мл концентрованої хлоридної кислоти. При інтенсивному протіканні реакції проводять охолодження на водяній бані. Після додання хлоридної кислоти колбу періодично струшують і нагрівають на водяній бані протягом 30 хв. Відсутність запаху гіркого мигдалю, характерного для нітробензолу, свідчить про закінчення реакції відновлення.
Гарячу суміш обережно нейтралізують 40 %-им розчином натрій гідроксиду до лужної реакції середовища за червоним лакмусовим папером. Анілін відганяють з водяною парою (до прозорого розчину).
Виділення продукту. До отриманого розчину додають насичений розчин натрій хлориду (на 100 мл розчину беруть 20 г солі) і відділяють анілін в ділильній лійці.
Анілін, який залишився у водному розчині, екстрагують етером (трьома порціями по 10 мл). Етерну витяжку додають до аніліну, висушують гранульованим натрій гідроксидом і відганяють етер. Потім в колбу вносять цинковий пил, відганяють анілін з повітряним холодильником і відбирають фракцію з Ткип = 182 – 184 ºС.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.22.
Характеритиска кінцевого продукту. Анілін – найпростіший ароматичний амін, безбарвна рідина з характерним запахом. Змішується у всіх співвідношеннях зі спиртом, етером, бензолом; розчинний в більшості органічних розчинників; Тпл = – 6,15 °С, Ткип = 184 °С, d420 = 1,0268, nd20 = = 1,5863.
Токсичність. Перетворює гемоглобін в метгемоглобін. Токсичний. ГДКр.з. = 0,1 мг/м3, ЛД50 = 550 мг/кг.
Використання. У виробництві барвників, вибухових речовин, проявників для фотографій, прискорювачів вулканізації каучуку, фармацевтичних препаратів та пестицидів.
5.8.2 Фенілгідроксиламін
Схема синтезу
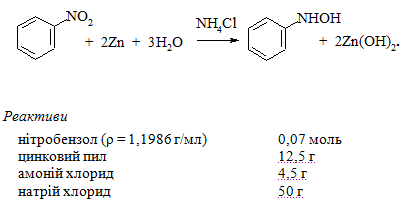
Методика синтезу. У фарфоровому стакані, оснащеному механіч-ною мішалкою і термометром, в 135 мл води розчиняють 4,5 г амоній хлориду і додають 0,07 моль нітробензолу. Протягом 15 хв до суміші при постійному перемішуванні поступово додають 12,5 г цинкового пилу. Якщо цинковий пил активний, температура суміші самовільно підвищується до 60 – 65 ºC. За необхідністю реакційну масу нагрівають до цієї температури на водяній бані. Після додання цинкового пилу вміст стакана перемішують протягом 15 хв до закінчення реакції, про що свідчить зникнення запаху нітробензолу і пониження температури реакційної суміші.
Теплий розчин фільтрують в конічну колбу об'ємом 200 мл через скляну лійку, осад промивають 20 мл гарячої води. Потім додають до фільтрату 50 г натрій хлориду і поміщають колбу в кристалізатор з охолоджувальною сумішшю (лід – натрій хлорид).
Виділення продукту. Фенілгідроксиламін виділяється у вигляді довгих світло-жовтих голок, які відфільтровують під вакуумом і сушать на фільтрувальному папері при температурі 40 – 50 ºC.
Довідкові, розрахункові та експериментальні дані заносять до таблиці 5.23.

Характеристика кінцевого продукту. Фенілгідроксиламін – безбарвна тверда речовина. Легко розчиняється в етері, спирті, хлороформі, гарячому бензолі, обмежено в воді; Тпл = 82 °С.
Використання. В органічному синтезі, для отримання пестицидів, фармхімпрепаратів.
Контрольні запитання
1. Яку реакцію називають реакцією відновлення? При протіканні яких реакцій відбувається відновлення органічних речовин?
2. До яких сполук відновлюються алкени, карбонові кислоти, естери, нітросполуки?
3. За яких умов відбуваються реакції гідрування алкенів, алкінів і аренів?
4. Як здійснити перетворення пропін – пропен – пропан? Наведіть схеми реакцій та зазначте умови їх протікання.
5. За яких умов відновлюються карбонові кислоти? Наведіть схему від-новлення оцтової кислоти.
6. Які продукти утворюються при відновленні етилацетату за Буво-Бланом? Наведіть рівняння реакції.
7. Наведіть схему реакції відновлення хлорангідриду оцтової кислоти за Роземундом. Який спирт утвориться при каталітичному гідруванні продукту цієї реакції?
8. У якому середовищі відновлення нітробензолу цинком відбувається через стадію утворення азобензолу? Наведіть схему реакції.
9. Наведіть схему реакції відновлення нітробензолу цинком в присутно-сті хлоридної кислоти.
10. Який продукт отримують відновленням нітробензолу цинком в при-сутності амоній хлориду? Наведіть схему реакції.