2.1.4 Хімічні властивості алканів
Алкани, у порівнянні із іншими класами органічних сполук, мають низьку реакційну здатність: при звичайних умовах не реагують із концент-рованою сульфатною кислотою, лугами та сильними окисниками. Це мож-на пояснити міцністю -зв’язків та їх схильністю до гомолітичного розще-плення. Таким чином, можна зробити деякі висновки щодо реакційної зда-тності алканів: – для цих сполук мають бути характерними реакції заміщення (роз-рив зв’язку С–Н) та реакції розщеплення (розрив зв’язку С–С); – ці реакції повинні проходити в досить жорстких умовах (t °C, P, h, ініціатор); – враховуючи низьку полярність зв’язків С–Н та С–С, для них пови-нен бути характерним гомолітичний розрив зв’язків з утворенням радика-лів. Галогенування алканів під дією галогенів проходить із утворенням моно- та полігалогенопохідних алканів. Загальна схема утворення монопохідних галогеноалканів: 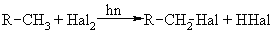 Швидкість реакції в залежності від природи алкану зменшується у ряду: третинний > вторинний > первинний, а в залежності від природи га-логену у наступній послідовності: F2 (з вибухом) > Cl2, Br2 (опромінювання світлом або t 300 °C) > > I2 (практично не взаємодіє без застосування спе-ціальних йодуючих агентів, наприклад, (СН3)3С–ОІ). Хлорування метану відбувається за схемою радикального заміщення (SR): 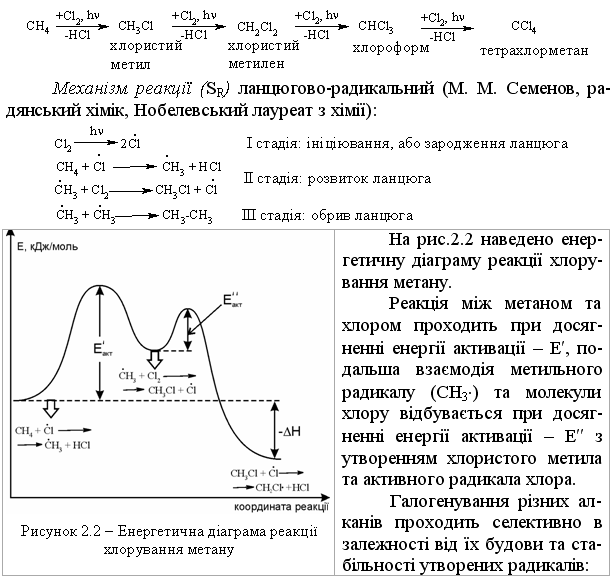 третинний > вторинний > первинний. При цьому їх термодинамічна ста-більність визначається енергією дисоціації зв’язку С–Н, тобто енергією утворення вільних радикалів та ефективністю делокалізації при цьому за-ряду радикального центру (табл. 2.3). 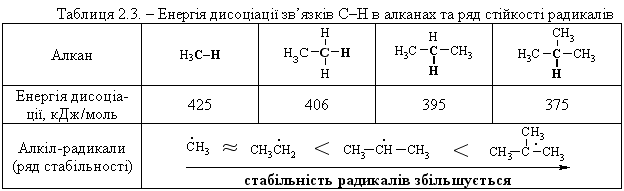 Можливість селективного галогенування закономірно пов’язана із природою галогену (Cl2, Br2) та швидкістю таких реакцій біля первинного ( ), вторинного ( ) та третинного ( ) атома карбону (табл. 2.4). 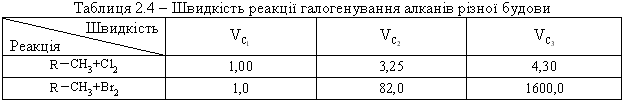 У випадку взаємодії (супряження) неспареного електрону із -електронною системою (алкени, арени) стабільність таких радикалів наба-гато збільшується: 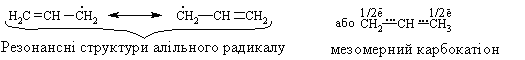 Кінетична стабільність радикалів зумовлена, в першу чергу, прос-торовими перешкодами, що впливає на їх реакційну здатність. Так, 4-метил-2,6-ди-трет-бутилфенол має об’ємні трет-бутильні ра-дикали, що знаходяться у о-положенні відносно гідроксильної групи. 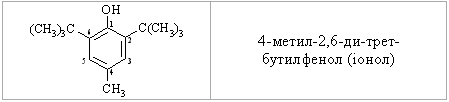 об’ємні третбутильні замісники екранують атом Оксигену та заважають перебігу радикальних реакцій по цьому реакційному центру. Виходячи із цього положення, іонол широко використовується у багатьох промислових сумішах (пластмаси, гуми, мастила, оливи тощо) як ефективний антиокси-дант. Розглянуті на прикладі реакції галогенування алканів теоретичні по-ложення (природа субстрата та реагента, стабільність радикалів) будуть характерними і для інших реакцій, що відбуваються за радикально-ланцюговим механізмом (SR). Нітрування (nitriding) алканів нітратною кислотою за звичайних умов не відбувається, а при нагріванні, як правило, іде їх окиснення. М. І. Коновалов, (1889 р.) встановив, що при нагріванні (~ 140 °С) під дією розведеної нітратної кислоти відбувається нітрування алканів за радикаль-ним механізмом (SR) із задовільним виходом кінцевих нітроалканів: 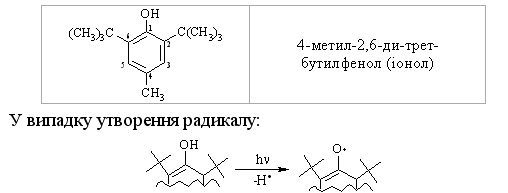 В таких умовах забезпечується селективність нітрування (ізопентан нітрується переважно по третинному атому карбону). Парофазне нітруван-ня (N2O4; t 400 °C) не забезпечує селективності процесу нітрування. 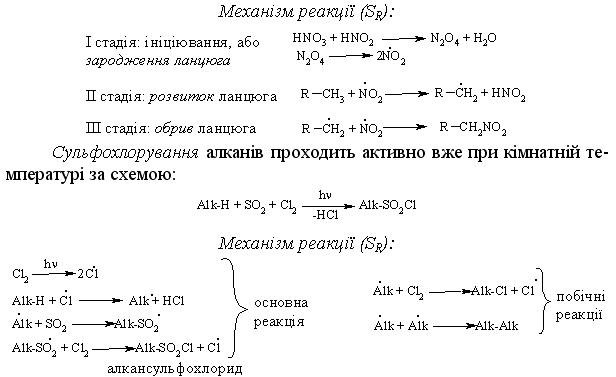 Реакцією сульфохлорування отримують суміш первинних та вторин-них алкансульфохлоридів. Третинні алкансульфохлориди, внаслідок прос-торових перешкод, не утворюються. Алкансульфохлориди є вихідними сполуками промислового вироб-ництва поверхнево-активних речовин (ПАР), які широко використовують як миючі засоби: 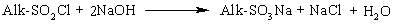 Окиснення. Алкани як паливо горять із виділенням великої кількості теплової енергії (повне окиснення): 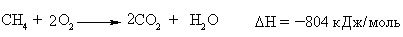 У випадку неповного окиснення (окисники: О2 повітря, KMnO4, K2Cr2O7; температура: ~150-200 °С; каталізатор: MnO2, MnAc2) утворю-ється суміш оксигенвмісних вуглеводнів: спиртів, альдегідів, кетонів та карбонових кислот. Механізм реакції радикально-ланцюговий (SR): 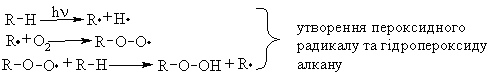 Стабільність гідропероксидів алканів в залежності від будови ради-калу R змінюється у ряду: третинний > вторинний > первинний. В залежності від будови гідропероксиду алкану ROOH утворюються різні продукти реакції. Первинні гідропероксиди при дисоціації зв’язку О–О утворюють ал-кокси-радикали, які далі окиснюються за схемою: 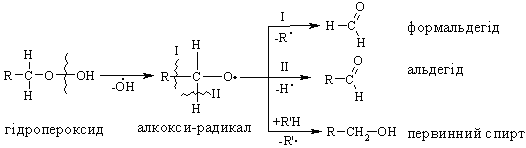 Вторинні гідропероксиди алканів утворюють при окисненні дещо інші продукти реакції: 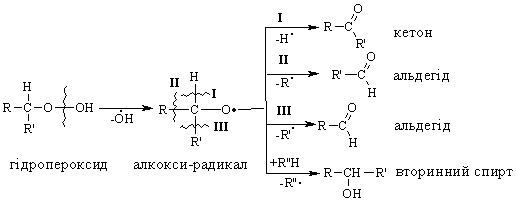 У випадку третинних гідропероксидів алканів реакція окиснення проходить за такою схемою: 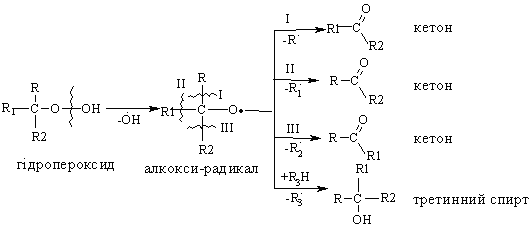 Особливості процесу окиснення: – гомолітичний розрив зв’язку С–С завжди проходить по -положенню відносно атома оксигену алкокси-радикалу (на схемах позна-чення І, ІІ, ІІІ); – спирти та альдегіди окиснюються далі, відповідно до кетонів або карбонових кислот. |